Junta Directiva de la Asociación por un Acceso Justo al Medicamento.
Parece razonable pensar que, en caso de pandemia, cuando toda la población mundial está amenazada, los medicamentos, vacunas, diagnósticos y otros productos sanitarios deberían ser accesibles a todas las personas en condiciones de igualdad. Los países deberían promover y financiar la investigación y el desarrollo de las medidas y productos sanitarios precisos, y se deberían ofrecer licencias no exclusivas a todos los fabricantes para lograr el máximo de producción en el menor tiempo posible, distribuyendo en todo el mundo estos productos con un precio que cubriera los costes de producción y sin añadir beneficios abusivos para ningún intermediario. De esta forma la pandemia afectaría a menos personas y de manera menos severa, y no dañaría tanto al conjunto de la economía y de la sociedad, en todos los países.
En noviembre de 2021, cuando las consecuencias de la pandemia de la COVID-19 estaban bien presentes en la sociedad mundial, y la falta de equidad en el acceso a las vacunas hería la conciencia de la humanidad, una sesión especial de la Asamblea Mundial de la Salud estableció el Grupo Intergubernamental para la Negociación de un Tratado de Pandemias. El 20 de mayo de 2025, después de más de tres años de negociaciones, la Asamblea Mundial de la Salud de la OMS ha aprobado este Tratado, de acuerdo con el Artículo 19 de la Constitución de la OMS, que regula los acuerdos vinculantes. (1)
Sin embargo, aunque el Tratado es vinculante y obliga a las partes a cumplir, lo cierto es que no recoge ninguna obligación. Es como si dijera: queda usted obligado a hacer lo que buenamente pueda y quiera. Así, el Artículo 9.5., sobre investigación y desarrollo, apunta a la inclusión de condiciones para las empresas que reciban financiación pública (concesión de licencias no exclusivas, precios asequibles, transferencia de tecnología y conocimiento…), pero no las hace obligatorias, sino que señala que los países podrán prever disposiciones en este sentido.
El Tratado no cumple las expectativas planteadas en 2021. Ya lo advertíamos en un editorial del pasado noviembre, en el número 34 de la Revista AJM. Durante la pandemia se evidenció que los países ricos acumulaban las vacunas, diagnósticos y tratamientos, mientras que los países del Sur Global quedaban relegados a las sobras. La causa de esta falta de equidad fue la concesión a determinadas empresas por parte de los países de monopolios para comercializar las vacunas y otros productos. En efecto, los países otorgan Derechos de Propiedad Intelectual (patentes, secretos comerciales, y otros), que permiten a las empresas decidir: dónde produzco, cuánto produzco, a quién vendo y a qué precio. En una situación de emergencia, como la pandemia, con una aplicación de las vacunas a miles de millones de personas, fijar precios diez o veinte veces por encima del coste, suponía un abuso que permitió generar unos beneficios de más de 100.000 millones de dólares. Se supone que los monopolios se conceden a las empresas para recuperar los costes de I+D. Pero en este caso, dichos costes los habían asumido los gobiernos con subvenciones y compras anticipadas. Fue un beneficio abusivo brutal. Y tuvo consecuencias devastadoras, al impedir la producción en plantas de todo el planeta y exigir precios que muchos países no podían pagar. (2)
Cuando se habló del Tratado de Pandemias para evitar que volviera a suceder la “catástrofe moral” de la que habló el director general de la OMS, pensábamos en un Tratado que incluyera la obligación de suspender automáticamente las patentes y otros derechos de propiedad intelectual relacionados con los productos sanitarios y medicamentos para la pandemia, y que estableciera la obligatoriedad de transferir la tecnología y el conocimiento necesarios para fabricar los diferentes productos. Además, se garantizaría una fabricación descentralizada en todo el planeta, para lo que se incrementarían los recursos de producción en las zonas menos desarrolladas. Y, por último, se garantizaría una distribución equitativa, a precio de coste de fabricación.
El Tratado aprobado el 20 de mayo en la Asamblea Mundial de la Salud no cumple esos objetivos (3). Si volviera a ocurrir una pandemia, que ocurrirá, la respuesta sería la misma. Dicho de otra manera, el monte parió un ratón.
El Tratado pretende mejorar la prevención, preparación y respuesta ante pandemias, sobre todo, garantizar la equidad en el acceso a vacunas, tratamientos y diagnósticos, así como a la atención sanitaria, pero no establece los mecanismos ni la financiación para poder garantizarlo. Es una pérdida de oportunidad difícilmente justificable.
Llaman la atención algunos aspectos. En primer lugar, la falta de consenso. En efecto, después de tres años de debates y con un texto nada ejecutivo, no se ha conseguido aprobarlo por consenso sino por votación. Por razones distintas (unos piensan que se quedó demasiado corto, otros que fue demasiado lejos), 57 países no han votado a favor. Así, según TWN, el tratado recibió 124 votos a favor y 11 abstenciones (entre éstos, Italia, Polonia, Países Bajos y Bulgaria), mientras otros 46 países estuvieron ausentes (como EEUU, cuyo presidente ha ordenado la retirada de esta Organización). (4)
En segundo lugar, la entrada en vigor. Aunque el Tratado se ha aprobado, no entrará todavía en vigor. En efecto, no está abierto a la ratificación de los países, y no lo estará hasta que se resuelva uno de los asuntos más espinosos, recogido en el Artículo 12: el Sistema de Acceso a los Patógenos y Participación en Beneficios (sistema PABS), mecanismo que debe regular y garantizar el acceso a “los materiales y la información sobre secuencias de los patógenos con potencial pandémico”, y que, al mismo tiempo, debe garantizar “la participación rápida, oportuna, justa y equitativa en los beneficios que se deriven del intercambio y/o la utilización de Materiales e Información sobre Secuencias PABS con fines de salud pública”. Este mecanismo se ha relegado a un Anexo que se discutirá en el próximo año. Para ello se crea un nuevo Grupo de Trabajo Intergubernamental (IGWG), que deberá reunirse a más tardar el 15 de julio de 2025, y redactar el Anexo para presentarlo a la 79ª Asamblea Mundial de la Salud en mayo de 2026.
Parte de los países que se abstuvieron o se ausentaron en la votación expresaron su disconformidad con que se aprobara el Tratado sin el Anexo del Artículo 12, que es “el alma” del mismo, y que condiciona su entrada en vigor. Las posiciones sobre cómo debe ser el sistema PABS no son coincidentes. Por ejemplo, Bangladesh insiste en que, con carácter obligatorio, se deberá garantizar acceso equitativo a vacunas y otros productos sanitarios, que se deberán garantizar licencias no exclusivas a los fabricantes en todas las regiones, así como transferencia de tecnología y conocimiento. Mientras Suiza reclama un enfoque voluntario y pragmático, teniendo en cuenta los puntos de vista de los fabricantes titulares de las patentes. Estas diferencias permiten pronosticar que el acuerdo sobre el Anexo no será fácil y que, quizá, podría dejar el Tratado en un limbo sin salida.
¿Por qué aprobarlo entonces ahora?
Parece que la razón principal es política: en un momento de tensión internacional, de auge de los nacionalismos y los discursos de exclusión, y de debilitamiento de la OMS con la decisión de EEUU de abandonarla, se trataría de poner en valor el mantenimiento de mecanismos multilaterales, el discurso de solidaridad y equidad, y el reconocimiento de la OMS en la coordinación de iniciativas de salud globales. Pero el problema es que esta aprobación no transmite una sensación de fortaleza para los principios de equidad y solidaridad, sino todo lo contrario.
Un ejemplo es el Artículo 12.6. Como el Tratado ha renunciado a exigir que, en caso de pandemia, se suspendan los Derechos de Propiedad Intelectual, se obligue a la transferencia de tecnología y conocimiento, se establezcan plataformas abiertas de innovación, etc., busca un mecanismo paliativo para poder apoyar a países de bajos ingresos. Así, establece que, durante una pandemia cada fabricante deberá poner a disposición de la OMS el 10% de su producción real, como una donación, más otro 10% disponible para ser adquirido a precios asequibles, previo contrato mutuamente acordado. ¿Estarán obligados todos los fabricantes, o será voluntario firmar esos contratos? ¿Se aplicará solo a los casos en los que hayan utilizado materiales de patógenos cedidos por un país? ¿Beneficiarán solo a esos países? ¿Serán transparentes estos contratos, o serán confidenciales como hasta ahora? Es poco probable que este sistema llegue a concretarse, por la falta de capacidad operativa de la OMS y de voluntad de las empresas. Si llegara a concretarse, ¿cómo se distribuirían esos productos?
Para Germán Velásquez la cesión prevista en el Instrumento PABS “no constituye una solución sostenible, sino más bien una institucionalización de la inequidad”. “El PABS debería aspirar a garantizar un acceso justo y universal a los recursos sanitarios, no a establecer cuotas asistenciales sujetas a condiciones”. El antecedente del mecanismo COVAX durante la pandemia de la COVID-19 mostró la insuficiencia de estos mecanismos, promovidos por las grandes empresas como coartada para no ceder en la suspensión de patentes y transferencia de tecnología.
Un Tratado “sin dientes”
Germán Velásquez entiende que el documento aprobado no cumple con el mandato original que dio la Asamblea. “En lugar de un tratado internacional vinculante, mantiene la lógica de desigualdad, reproduce mecanismos voluntarios poco efectivos y refuerza los desequilibrios de poder existentes en la gobernanza sanitaria mundial”.
“La mayoría de los contenidos son indicaciones no vinculantes. No hay obligaciones jurídicas claras ni mecanismos de evaluación de cumplimiento”. “Lo esencial permanece sin resolver: garantizar el acceso equitativo y oportuno a medidas médicas durante las emergencias”. (5)
También Ebere Okereke es crítico respecto al Tratado: “Un tratado de pandemias sin dientes, dejará a África y al mundo expuestos”. Y añade: “Sin mecanismos de cumplimiento obligatorio, aun los mejores compromisos pueden quedarse en meras aspiraciones morales”. (6)
Por su parte, Mark Eccleston-Turner, Michelle Rourke y Stephanie Switzer, escribieron en Think Global Health el pasado 20 de mayo: “Los derechos de propiedad intelectual han sido el elefante en la habitación durante las negociaciones, pero el texto final es mudo sobre esta cuestión”. “El Tratado tiene objetivos laudables, pero parece poco aplicable. Queda pendiente de la discusión del Anexo de los PABS”. (7)
Ellen ‘t Hoen recuerda que en diciembre de 2021 cuando se comenzó a discutir el Tratado, “la atmósfera era de ambición y solidaridad. Sin embargo, esta se evaporó rápidamente a partir del primer borrador”.
“La Transferencia de Tecnología y los asuntos relacionados con los derechos de propiedad intelectual suscitaron importantes discrepancias. Los países ricos, singularmente la Unión Europea, defendían que debían ser decisiones voluntarias de las empresas y países, según acuerdos mutuamente acordados” (8). Y, como se ha visto, lograron imponer su criterio, en función de los intereses de la BigPharma.
En definitiva, el Tratado no aborda de forma clara y eficaz asuntos clave. No hay compromisos obligatorios en el acceso equitativo a las medidas médicas y productos sanitarios, ni tampoco en la transferencia de tecnología y conocimiento.
Transferencia de tecnología y conocimiento y financiación
En este sentido, el Artículo 11, sobre Transferencia de Tecnología y cooperación en relación con el conocimiento para la producción de productos relacionados con la pandemia, establece que cada país promoverá la transferencia de tecnología y conocimiento, “mutuamente acordada”, es decir, voluntaria. El Tratado fracasa en su intención de ir más allá. Solamente reconoce que los países podrán aplicar las flexibilidades del acuerdo sobre los Aspectos de los Derechos de Propiedad Intelectual Relacionados con el Comercio, ADPIC, como licencias obligatorias y otras. No deja de sorprender que, como señala Ellen ‘t Hoen, hasta el último momento algunos países de altos ingresos insistían en que en todos los casos la transferencia de tecnología debería ser voluntaria, anulando así, incluso, las flexibilidades de los ADPIC.
En todo caso, la transferencia voluntaria, como se demostró en la pandemia de la COVID-19 con la Health Technology Access Pool de la OMS, no funcionó. Y las flexibilidades de los ADPIC no son suficientemente ágiles (y tienen que superar demasiadas barreras jurídicas y políticas) para ser operativas en una pandemia.
El Artículo 13 se refiere a la Red de Suministro Global y Logística. Se crea esta Red para facilitar el acceso seguro, rápido y equitativo a medidas relacionadas con la pandemia. Se desarrollará por la OMS en colaboración con los países. La Conferencia de las Partes deberá desarrollar este mecanismo (estructura, funciones, etc.). Si llega a desarrollarse puede ser una buena herramienta. Pero estará vacía porque no se ha incluido en el Tratado la suspensión automática de los monopolios en caso de pandemia.
Resumiendo, el Tratado revisa todos los aspectos relativos a la prevención, preparación y respuesta ante las pandemias. Pero expresa buenas intenciones, no mandatos. Son indicaciones a los países de lo que deberían hacer (reforzar la vigilancia, aumentar los recursos humanos en salud, cooperación en investigación, establecer y aumentar la capacidad de producción de contra-medidas, concesión de licencias no exclusivas, etc.). Pero no obliga a los Estados, ni fija calendarios, ni ofrece recursos.
En efecto, según su enunciado, el Artículo 18 se refiere a la financiación sostenible.
Sin embargo, no hay mecanismos de financiación concretos y predecibles. Se dice que “las Partes reforzarán una financiación sostenible y predecible, de una forma transparente”. Cada País, de acuerdo con su capacidad y disponibilidad de recursos, mantendrá o incrementará sus fondos nacionales para prevención, preparación y respuesta a pandemias. Y movilizará financiación adicional para países de menos recursos. Pero no se cuantifica, ni se hace obligatoria esta financiación, por ejemplo, para reforzar los servicios de salud, o para promover plataformas tecnológicas para la fabricación de vacunas y otros productos sanitarios.
En cuanto a la financiación de la implementación del Tratado, se crea un Mecanismo de Financiación Coordinado. Desde aquí se supone que se reforzarán capacidades para prevenir, preparar y responder ante pandemias, sobre todo en países en desarrollo.
No se concretan cantidades y se deja abierto a su desarrollo en la Conferencia de Partes.
Monopolios, patentes y otros derechos de propiedad intelectual
En el preámbulo del Tratado se sigue asumiendo la narrativa de la industria farmacéutica, reflejando el enorme peso de esta en los gobiernos de una mayoría de países: “Reconociendo que la protección de la propiedad intelectual es importante para el desarrollo de nuevos medicamentos y reconociendo las preocupaciones sobre sus efectos en los precios, … y recordando que el Acuerdo … sobre los ADPIC … ofrece flexibilidad para proteger la salud pública…”
Los derechos de propiedad intelectual, patentes, secretos comerciales y otros, son tremendamente dañinos en el campo de la salud. No facilitan el descubrimiento y desarrollo de nuevos medicamentos. Permiten la creación de monopolios que tienen la potestad de fijar precios astronómicos sobre medicamentos esenciales, como las vacunas en caso de pandemia. Las “flexibilidades” del Acuerdo sobre los ADPIC no han sido capaces de frenar esa sangría sostenida sobre los sistemas de salud de todo el mundo. Que los Derechos de Propiedad Intelectual y sus monopolios sirven para promover la innovación es un engaño. Una gran mentira, repetida y repetida. La mayor parte de la investigación innovadora (como ocurrió con las vacunas en la COVID-19) tiene financiación pública directa. El resto de la investigación innovadora, que paga la industria, lo hace con el dinero que le dan los Servicios de Salud y los pacientes a través de los sobre precios que fijan gracias al monopolio. Pero los gobiernos no controlan si esa empresa ya ha cubierto los costes de investigación, más un beneficio razonable, y permiten fijar precios abusivos, 10, o 100 veces por encima de los costes de fabricación y de I+D, en lugar de suspender el monopolio.
Si después de una pandemia no hemos sido capaces de lograr una mayoría de países que cambien este sistema de abuso, no será fácil hacerlo para otros productos y en tiempos de normalidad. Pero nos va la vida en ello. Porque la presión de los precios de nuevos medicamentos sobre los pacientes y los Sistemas de Salud sigue creciendo, y llegará un punto que los acabe desangrando. Por eso hemos de seguir insistiendo en cambiar el modo de financiar la I+D, prohibiendo los monopolios en medicamentos, así como todos los instrumentos jurídicos que los sustentan.
Se ha perdido una gran oportunidad. Habrá que seguir luchando.
Este número 38 recoge las alegaciones completas presentadas por AAJM al anteproyecto de Ley de Medicamentos. Desde la Asociación se considera que los medicamentos y los productos sanitarios deben obedecer a la consideración de bienes sociales de utilidad pública y, en ese sentido, deben tener una prelación absoluta. Algunos de las mejoras propuestas al anteproyecto son reducir los copagos hasta eliminarlos, impulsar la fabricación pública de medicamentos, así como la opción a su distribución directa desde los centros de salud. Esta publicación, en relación con el citado anteproyecto, aporta asimismo el editorial, de Patricia Lacruz Gimeno, miembro de la Comisión Editorial, que considera que el anteproyecto “responde a una estrategia dirigida por la industria farmacéutica”; un análisis del texto de Ángel Mª Martín Fernández-Gallardo, vicepresidente de la AAJM y exjefe del Área de Farmacia del SESCAM, que estima que los fabricantes de medicamentos “confunden o engañan a la sociedad con un descaro que sólo es posible por la seguridad que tienen de controlar perfectamente el discurso y de que los medios afines retransmiten y amplifican sin la más mínima crítica, verificación ni contraste”; y otro articulo más, referido al mismo asunto, de Fernando Magro, ex-director general del INSALUD, que concluye indicando que “queda camino por recorrer en estos momentos claves que deben dirigirse a una mayor unidad europea que no sea meramente formal, sino real y ejecutiva, política, económica y cultural, para que el medicamento también sea un elemento de referencia, de uso, de consumo de accesibilidad igual para todos”.
Entre los originales, hay, igualmente, una valoración de Fernando Lamata, psiquiatra y presidente de la Comisión Editorial, sobre el Plan de Acción de Salud Mental 2025-2027, que fue aprobado en el Consejo Interterritorial del Sistema Nacional de Salud el pasado 4 de abril, del que señala que “las líneas estratégicas que recogen los diferentes objetivos son muy acertadas. Nos dicen lo que hay que hacer. Sin embargo, las acciones que se definen para lograr dichos objetivos se quedan muy cortas”.
En otras fuentes, el Nº 38de la rAJM, se detalla una hipótesis formulada por Dean Baker, de CEPR, que viene a indicar que la política de aranceles de Trump, puede suponer que le salga el tiro por la culata, dada su transgresión del libre comercio, y que el resto de países no respeten los monopolios estadounidenses de patentes y derechos de autor, con lo que alcanzarían ahorros muy sustanciales. También este número se hace eco de un interesante, descriptivo y claro artículo de Salud por Derecho donde, entre otros temas, se abordan algunas de las propuestas relacionadas con el Tratado de Pandemias de la Organización Mundial de la Salud, relacionadas con la transferencia de tecnología y el sistema de acceso y reparto de beneficios. Finalmente, desde otras fuentes, se aporta un análisis de Sara Plaza Casares, de El Salto, sobre el anteproyecto de Ley de Medicamentos, en el que recogen varias opiniones destacadas y, entre ellas, las del vicepresidente de la AAJM, Ángel Mª Martín, que subraya que “necesitamos un sistema que produzca una bajada real de los genéricos y no una bajada estética. Además, que no se cargue la imagen de los genéricos, que tanto ha costado reforzar. Si el Ministerio da distintos precios a unos y a otros, ¿quién se va a creer que es lo mismo?”
Fue en el segundo cuatrimestre del año 2020 cuando el Ministerio de Sanidad comenzó a elaborar el Plan de Recuperación, Transformación y Resiliencia (PRTR) y centró las reformas e inversiones a desarrollar en los próximos años en nuestro sistema sanitario público. Así pues, las plasmó en el componente 18 denominado “Renovación y ampliación de las capacidades del Sistema Nacional de Salud”, que contiene 5 reformas y 6 inversiones con un presupuesto estimado de 1.169 millones de euros.
Las reformas recogidas son todas ambiciosas y muy necesarias y una de las 5 es la reforma de la Ley de Garantías y Uso Racional del Medicamento, cuya consulta previa se realizó en julio de 2022.
Es posible que muchos no lo advirtieran, pero el PRTR no contenía de forma original la estrategia de la industria farmacéutica, al menos en el componente 18. De hecho, como se puede observar, no tiene hito asociado. Al respecto, el PRTR el mismo día de la publicación si mal no recuerdo, fue modificado incluyéndose esta estrategia como parte de las reformas del Sistema Nacional de Salud (SNS).
Personalmente, debo expresar, una vez más y cuantas tantas hagan falta, que no estoy de acuerdo en que la citada estrategia se enmarque en el eje que persigue fortalecer al SNS y menos que la lidere el Ministerio de Sanidad. Y esto no es una cuestión hueca porque, desde mi perspectiva, tiene consecuencias importantes. Para mí, la principal, es que expone (más aún de lo que está) a nuestro sistema sanitario público al interés económico de las farmacéuticas.
Para contextualizar mi opinión, es oportuno compartir más información. Allá voy. En las primeras reuniones, convocadas por el Ministerio de Industria, que se mantuvieron junto con Sanidad y Ciencia, abordamos la necesidad de disponer de la estrategia, cuya idea primigenia, por si hubiera alguna duda, fue puesta encima de la mesa por el sector farma. En estas, ni se trató su inclusión en el PRTR. Desde Sanidad trasladamos reiteradamente que debía liderarse por Industria. ¿Por qué? Por una simple cuestión de competencias y, fundamentalmente, de propósito: ¿qué queremos conseguir?
Antes de avanzar en esta reflexión, quiero poner de manifiesto varias cuestiones:
Considero que una estrategia que consolide y potencie el sector farma en nuestro país, fortaleciendo sobre todo la industria nacional, es necesaria.
Es el Ministerio de Industria el competente en la propuesta y ejecución de la política del Gobierno en materia de industria.
El PRTR contiene un componente específico para “impulsar la modernización y la productividad del ecosistema español de industria-servicios”. En concreto, es el 12. Política Industrial España 2030. Este componente tiene como reto fundamental “reforzar el peso de la industria en la economía española y aumentar la dimensión de las empresas industriales”. En esta se enmarcan reformas e inversiones concretas para sectores estratégicos estableciéndose como tal el sector salud, automoción, turismo, comercio y agroalimentario. Solo se nombra una vez la “industria farmacéutica”, pero no aparece por ninguna parte nada relacionado con el sector farma.
El Ministerio de Sanidad, en materia de medicamentos, tiene la competencia para el desarrollo y ejecución de la política farmacéutica, así como las funciones relativas a la financiación pública y fijación del precio de medicamentos y productos sanitarios.
Entonces, dado que el tema competencial no tiene discusión, ¿por qué está en el componente 18 y por qué la lidera el Ministerio de Sanidad? Esa es la pregunta que nos podríamos hacer.
Mi hipótesis, como antes he comentado, es el propósito que se persigue y quién o qué lo mueve. Dado el momento actual, debemos analizar, aunque sea muy por encima, la estructura de la publicada Estrategia de la Industria Farmacéutica 2024-2028.
Se articula en tres ejes: 1) el acceso de los pacientes, la cobertura de necesidades médicas no cubiertas y la sostenibilidad del SNS; 2) el fomento de la investigación, la innovación y el desarrollo; 3) competitividad, resiliencia y ecosostenibilidad del ecosistema industrial y sus cadenas de suministro.
El eje 1 tiene 12 líneas de acción, que representan más de la mitad de las contenidas en la estrategia, y éstas apuntan directamente a la línea de flotación de la política farmacéutica, en concreto al proceso de financiación y fijación de los precios de los medicamentos enmascaradas con el “acceso” de los pacientes a los medicamentos. Esto ya nos orienta sobre lo que se pretende. Si además relacionamos la estrategia con el anteproyecto de Ley de los medicamentos y productos sanitarios, actualmente en fase de información pública, vemos referencias incluso en esta última, donde en el propio preámbulo se destaca que “esta ley da respuesta no solo a la propia necesidad de reforma de la misma, sino que también forma parte de la implementación de esta Estrategia de la Industria Farmacéutica”.
Concluyendo, en mi opinión la estrategia, principalmente, está dirigida por la industria farmacéutica en un foro que han conseguido crear, privilegiado, por cierto, con un poder de toma de decisión que no debería haberse cedido a aquellos que tienen tal conflicto de interés, donde han logrado que además lo lidere Sanidad. Bien es cierto que uno tiene el poder que le dan.
Es un tema complejo: sí, nadie lo pone en duda. Pero es esencial para nuestro SNS que el enfoque de las soluciones que se definan para abordar los retos y los desafíos actuales y futuros tengan un propósito claro e inequívoco cuyo principal activo sea el SNS y que éste sea el que guie, siempre, las decisiones.
El Consejo Interterritorial del Sistema Nacional de Salud del pasado día 4 de abril aprobó el Plan de Acción de Salud Mental 2025-2027 (1).
Las líneas estratégicas que recogen los diferentes objetivos son muy acertadas. Nos dicen lo que hay que hacer (2). Sin embargo, las acciones que se definen para lograr dichos objetivos se quedan muy cortas. Y lo más importante: no se prevé la dotación de recursos para alcanzar los objetivos declarados. Por ejemplo, la primera línea, que impacta en todas las demás, es el refuerzo de recursos humanos en salud mental. Los objetivos son aumentar el número de profesionales en salud mental, mejorar sus condiciones laborales, garantizar su disponibilidad y retener el talento. Las acciones para alcanzar esos objetivos incluyen planificar la oferta de profesionales (formación), impulsar el acceso a la psicoterapia, promover el reconocimiento de la especialidad en Psicología Clínica de la Infancia y la Adolescencia; promover el apoyo mutuo entre pares, y elaborar informes que evalúen las necesidades de equipos y profesionales en salud mental. Son acciones interesantes, pero muy insuficientes para lograr los objetivos propuestos.
En efecto, las carencias actuales en la atención a problemas de salud mental son graves. En los últimos 12 meses, las personas que tuvieron que consultar por un problema de salud mental y lograron ser atendidas por un psiquiatra o psicólogo, lo fueron en un 62,5% en la sanidad privada y solamente un 37,5% en la pública (3). Esto refleja la falta de respuesta suficiente. De la misma forma, un 64,6% tuvo que esperar más de un mes a que le atendiera un profesional de psiquiatría o psicología (3). En ese tiempo, el problema de salud puede agravarse y, por eso, quien tiene posibilidad, recurre en muchas ocasiones a la sanidad privada. Otro dato que refleja la insuficiencia de los programas de promoción de la salud y prevención de la enfermedad es que entre 2016 y 2022 ha aumentado un 35% la prevalencia, ajustada por edad, de problemas de trastornos mentales y del comportamiento (4).
El número de psiquiatras en España, en relación con la población, es mucho menor que países de nuestro entorno. Debería aumentar de 13 / 100.000 habitantes a más de 20 / 100.000 habitantes, como en Alemania, Francia, Irlanda, Italia u Holanda. Unos 4.300 profesionales más (5). Lo mismo ocurre con los profesionales de psicología clínica, que deberían pasar de 6 a más de 20 /100.000 habitantes, unos 7.000 más. También en enfermería de salud mental se precisan 10.000 profesionales más, en Trabajo Social 6.000 profesionales más y otro tanto en personal administrativo. De igual manera, se debería contratar a 2.000 personas con experiencia propia que presten apoyo mutuo. El Plan debería partir de las estimaciones actuales más fiables, y trazar un calendario de dotación, con financiación finalista y cobertura inmediata y progresiva. Mientras tanto, como dice el Plan, se pueden hacer informes que recojan el detalle de todos los recursos actuales y de las necesidades más precisas por ámbitos geográficos, funciones, etc. Pero el problema de falta de atención y mala calidad de atención, por sobre carga de los servicios, es urgente. Esta situación se agrava además por la situación de deterioro asistencial de la Atención Primaria que impide a sus profesionales dedicar el tiempo necesario para detectar los problemas de salud mental de sus pacientes.
El Gobierno debe aprobar una financiación finalista suficiente para aumentar la dotación de las plazas necesarias y fijar un calendario de cobertura para los próximos 2-6 años.
En las demás líneas, todas ellas acertadas, ocurre lo mismo. No se cuantifican los recursos necesarios, la financiación necesaria y su aprobación por el Gobierno. La línea 2 se refiere a reforzar la salud mental comunitaria y las alternativas a la institucionalización. Sin embargo, en los últimos años estamos yendo en la dirección contraria. Para revertir ese proceso hacen falta recursos en la comunidad, capacidad de coordinación y recursos humanos sobre el terreno. La línea 3 pretende impulsar un modelo de atención orientado a los derechos humanos, que reduzca y limite al mínimo el uso de la contención mecánica y las intervenciones involuntarias. Pero, para lograrlo se necesita aumentar significativamente la dotación de personal, lo no se concreta en el Plan. La línea 4 pretende un uso racional de psicofármacos. En AAJM venimos defendiendo este objetivo desde hace años. El Plan impulsará la elaboración de una Guía de Práctica Clínica, lo cual está muy bien. Pero el uso inadecuado y la sobre prescripción (se estima que alrededor de un 20% de la población mayor de 18 años consume psicofármacos habitualmente) se deben en buena parte a la falta de tiempo suficiente de los profesionales, tanto en Atención Primaria como en Salud Mental. Para llevar a cabo programas de prevención, promoción de la salud, psicoterapias y otro tipo de intervenciones comunitarias que reduzcan el recurso a las pastillas se necesita personal. Al mismo tiempo, se debe reducir la presencia de la industria en la formación continuada, y reducir los precios de los nuevos medicamentos. Solo con el aumento de gasto farmacéutico público que se ha producido en 2024 en el SNS (1.696,4 millones de euros), se podrían financiar las plazas de 20.000 profesionales en salud mental (6). Pero es que el exceso anual de gasto farmacéutico público innecesario supera los 11.000 millones de euros, que se deberían destinar a este y a otros Planes de Salud.
A continuación, observamos como la línea 5, define el abordaje de problemas en colectivos de mayor vulnerabilidad, la línea 6, el impulso en la mejora de la atención a la salud mental perinatal, en la infancia y la adolescencia, la línea 7, la potenciación de los sistemas de información en salud mental y finalmente, la línea 8 la promoción de la salud mental en el trabajo. Todas las líneas anteriormente enumeradas del Plan de Acción son sin duda útiles e importantes. Pero de nuevo son muy insuficientes en cuanto a la dotación de recursos humanos y a la puesta en marcha de programas y dispositivos comunitarios.
El Plan no trasmite al Gobierno, ni a la sociedad, la urgencia del problema, ni tampoco dimensiona los recursos necesarios. La situación de la atención a la salud mental se está deteriorando día a día. Se precisa duplicar los recursos públicos destinados a salud mental para situarnos en la media de gasto público por habitante de la UE y responder adecuadamente a las necesidades de la población. Se precisa reducir el gasto farmacéutico y el gasto hospitalario en salud mental y aumentar los programas comunitarios. Por eso necesitamos un Plan con medios adecuados, mecanismo de financiación finalista, estándares de calidad y servicio, calendario de ejecución y sistema de evaluación que garantice una atención sanitaria de calidad y respetuosa con los derechos humanos.
Vicepresidente de la AAJM. Ex Jefe de Área de Farmacia del Sescam.
El sistema de precios seleccionados
En el artículo 116 del Anteproyecto de Ley de los Medicamentos y Productos Sanitarios se presenta como una gran novedad, para aumentar la competencia y bajar los precios de los medicamentos genéricos, una medida: el sistema de precios seleccionados. Sin embargo, este sistema ya está regulado y vigente desde 2012 cuando se añadió mediante un nuevo artículo, 93 bis, a la Ley de Garantías y Uso Racional de los Medicamentos en el Real Decreto-ley 16/2012, de medidas urgentes para garantizar la sostenibilidad del Sistema Nacional de Salud y mejorar la calidad y seguridad de sus prestaciones, pero que nunca se ha usado ni se ha tenido la más mínima intención de utilizar. Una medida que de haber decidido implementar realmente habría generado una auténtica bajada de los precios de estos medicamentos por:
La obligación que impone la Ley actual de que se dispense obligatoriamente el medicamento de precio más bajo.
Porque el precio seleccionado marca el precio más bajo durante dos años.
Y porque, durante el tiempo de vigencia del precio seleccionado, quedan excluidas de la financiación pública las presentaciones que no resulten seleccionadas.
Pero esa medida fue una herramienta de distracción que el PP incluyó de farol en plena crisis económica, cuando los “hombres de negro de Bruselas” exigían fuertes recortes y no sólo medidas estéticas para reducir el gasto farmacéutico galopante en España. Desde entonces duerme el sueño de los justos con el beneplácito del Ministerio y para gozo y tranquilidad de la industria farmacéutica. Y nunca se ha desarrollado (seguramente se incluyó con la complicidad de la industria farmacéutica), motivo por el cual no se ha puesto en marcha).
Sin embargo de nuevo se reinventa, muy descafeinado, en el Anteproyecto que ha salido a información pública, en el cual:
Desaparece la obligación de que se dispense obligatoriamente el medicamento de precio más bajo.
El precio seleccionado deja de ser un precio único y se convierte en una horquilla de precios, en el que el precio menor será uno más de los seleccionados, y al que le va a resultar muy difícil ganar una cuota de mercado muy diferente a la del resto más caros. Y, sin ese incentivo, es difícil que algún laboratorio apueste por una bajada poco más que testimonial.
Y, como guinda, se permite a las marcas que quieran mantener sus precios por encima del precio de referencia seguir financiadas, algo que la Ley actual no permite, y que es como soltar al lobo alrededor del rebaño de los seleccionados.
La rebelión en la granja
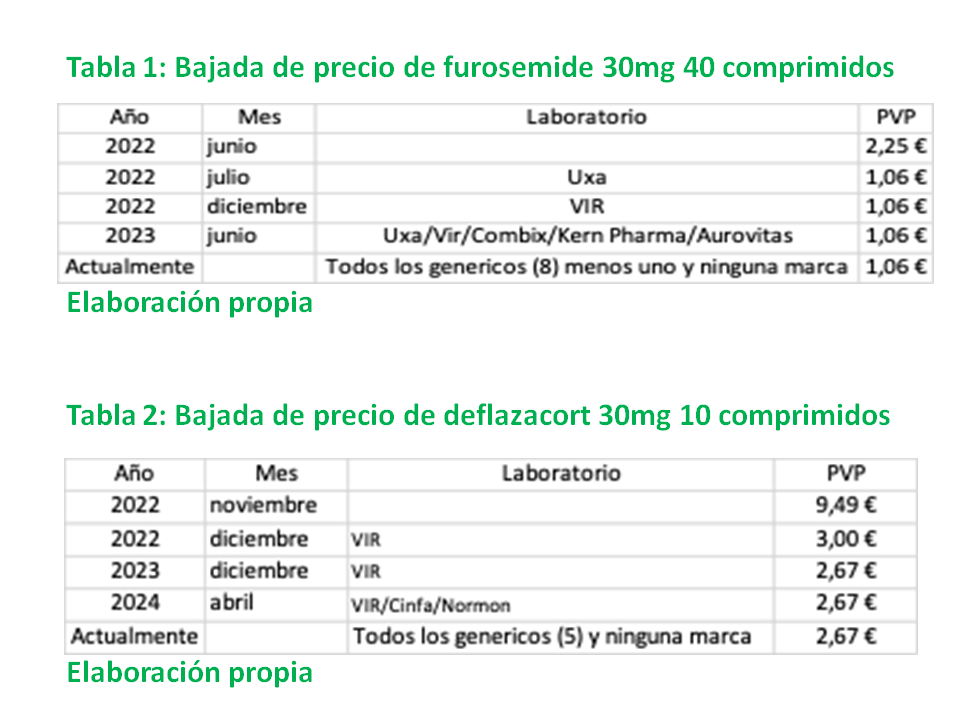
En julio de 2022, un laboratorio de genéricos, harto de ser el paria de la industria farmacéutica dio un golpe maestro: bajó el precio de la furosemida a menos de la mitad y sus medicamentos se convirtieron, de la noche a la mañana, en los únicos que según la Ley se deben dispensar obligatoriamente en toda España. Posteriormente, en diciembre de 2022, otro laboratorio se unió al rebelde igualando el precio de la furosemida y redoblando el desafío al bajar el precio de deflazacort a un tercio del precio que tenía hasta ese momento. De inmediato, el resto de laboratorios y la llamada “prensa del sector”, su coro repetidor de frases y slogan, inician una campaña para advertirnos del apocalipsis de desabastecimientos que se avecinaba ante tales desafíos. Y el resto de laboratorios de genéricos, que, según la Ley, podían haber bajado sus precios para igualar los de los farmacéuticas rebeldes en ese mismo mes, o en cualquiera de los meses siguientes, deciden no hacerlo y mantienen la estrategia del avestruz: cerrar los ojos o mirar para otro lado esperando que su profecía se cumpla y se desabasteciera el mercado español de furosemida y deflazacort para poder criticarlos y también al Ministerio por haber autorizado esa bajada de precios que califican de “temeraria”. Esperan y esperan, pero el mercado no se desabastece.
No se desabastece porque, para autorizar esas bajadas de precios, el Ministerio les había exigido que demostrasen que podían mantener ellos solos el abastecimiento completo del mercado nacional y así lo hicieron y cumplieron. Finalmente, pasado un año desde el inicio de la rebelión, y ante la evidencia de que con su estrategia habían regalado el mercado de furosemida y deflazacort a los rebeldes, algunas empresas farmacéuticas bajaron e igualaron el precio de sus medicamentos. Pero uno de ellos, ante el éxito cosechado, decide inmediatamente, en diciembre de 2023, una nueva bajada del precio del deflazacort. Ante este nuevo desafío, ¿qué hacen los anunciadores del apocalipsis? Se lo pueden imaginar, no esperar otro año sino bajar el precio del suyo.
Como resultado de esa rebelión, para esos dos principios activos el porcentaje de consumo de genéricos se acerca al 100% y las marcas se han batido en retirada, tal como no se cansan de repetir quiere la Asociación Española de Medicamentos Genéricos (AESEG). Además, los pacientes y el SNS se han beneficiado de importantes ahorros en sus bolsillos. ¿Y ha habido desabastecimientos? Pregunten a los coros de la prensa del sector.
En las Tablas 1 y 2 se muestra la secuencia temporal de las bajadas de precios de estos medicamentos.
Los interinos
Y, en plena rebelión, un día, a finales de 2023, aparecen en el Ministerio dos nuevos interinos (dicho con respeto, es una expresión que empleaba un bedel del Ministerio para referirse a los nuevos ministros y altos cargos, que llegaban con cada cambio de gobierno), con una limitadísima experiencia y conocimiento en el laberíntico campo legislativo del medicamento. Un complejo sudoku que domina a la perfección la industria farmacéutica, con sus asesores legales, sus economistas de la salud y su ejército de ex altos cargos y ex técnicos del Ministerio de Sanidad que tienen en nómina, que tardan dos telediarios en convencerles del mal sistema de precios seleccionados que les acaban de colar en el Anteproyecto de Ley del Medicamento. Sorprende además que los actuales responsables del Ministerio de Sanidad presenten a la sociedad, con entusiasmo, este sistema de precios como una novedad y un gran avance para bajar el precio de los genéricos.
Pero no, no lo es Sra. ministra y Sr. secretario de Estado, ni es una novedad ni es un avance. Son bridas que la industria farmacéutica ha introducido en el anteproyecto de Ley para que no les vuelva a ocurrir lo que pasó con furosemida y deflazacort, que amenazaba con extenderse y revolucionar el mercado de los genéricos con nuevos desafíos (en el verano de 2024, otro laboratorio se había unido a la rebelión con un nuevo principio activo). Pero bastó su anuncio, muy temprano, de que iban a cambiar el sistema para que ninguno más se haya atrevido a desafiar a los que imponen el statu quo ¿Quién se va a arriesgar para que a mitad de la partida le cambien las reglas para que le puedan dar jaque mate? Y Uds. ahora, en lugar de potenciar la incipiente revolución en la granja, nos presentan un anteproyecto para meter a los rebeldes en el redil y devolverlos a su anterior status.
El regreso al pasado
Pero esto no pasaría de ser una anécdota, o una película de intriga entre bandas rivales, si no fuera porque los espectadores, en este caso los pacientes y el SNS, son los pagadores finales de los sobreprecios artificialmente elevados que imponen, también a los medicamentos genéricos.
Con la nueva Ley y el regresivo sistema de precios seleccionados que configura, se consolidan los precios e incluso incrementan, al permitir que medicamentos genéricos idénticos se puedan vender a precios diferentes sin la más mínima ventaja para el paciente y que se mantengan en la financiación por encima del precio de referencia de las marcas, a las que además se les financiará una parte del precio. El efecto demoledor para la credibilidad del uso racional del medicamento que tienen esas dos medidas seguro que. no se les escapa, porque han sido testigos, como clínicos en ejercicio, de los años que ha costado convencer a la población de que un genérico y su marca son idénticos. Un convencimiento que sin duda reafirmó el hecho de que finalmente cuesten lo mismo; porque ¿quién se puede creer que, si el Ministerio autoriza a uno un precio de 10 y a otro un precio de 20, son idénticos? Permítanme un refrán popular: Ni el que asó la manteca con el dedo .
Sra. ministra, Sr secretario de Estado, con esta modificación legislativa nos devuelven a la prehistoria del uso racional del medicamento y van a permitir un nuevo asalto de la industria farmacéutica al bolsillo de los ciudadanos, que van a pagar más por lo mismo para que, supuestamente, mejore una estadística, la del consumo de genéricos.
Una propuesta claudicante
Basta leer el comunicado conjunto que sobre el Anteproyecto de Ley de Medicamentos y Productos Sanitarios han firmado, entre otros, Farmaindustria, la AESEG y la Asociación Española de Medicamentos Biosimilares (BIOSIM), para comprender las lágrimas de cocodrilo con las que lo han publicado. No piden que se elimine el sistema de precios seleccionados que regula la nueva Ley, sino “que su desarrollo regrese al camino que nos traza la Estrategia de la Industria Farmacéutica” (1). En resumen, quieren que una vez que han conseguido introducir en la Ley un sistema de precios seleccionados mucho más favorable a sus intereses que el vigente, (que, aunque nunca se haya desarrollado es una espada de Damocles para sus intereses) también quieren controlar el mando a distancia, porque, aunque la espada ahora sea de plástico, no sea que en “manos de irresponsables” le salten un ojo a algún laboratorio.
La patronal de los fabricantes de genéricos, AESEG, se queja de que la igualdad de precios, que desde 2011 impide superar un precio de referencia para los medicamentos con un mismo principio activo, ha menoscabado la razón de ser de los medicamentos genéricos, que se basan, precisamente, en la diferencia de precio (2). Pero se calla interesadamente que, con un mercado de genéricos con precios tan elevados, ninguno quiere renunciar, ni los laboratorios de marcas ni los de genéricos, a los ingentes beneficios que esos abultados precios generan. Y para ninguno de ellos bajar el precio es una opción. En eso están todos de acuerdo. En lo único en que discrepan es que las marcas quieren su parte del pastel y los genéricos no la quieren ceder, pero tampoco quieren bajar sus precios para que dejen de ser tan atractivos para las marcas. Y en esto llegan los nuevos inquilinos al Ministerio y, de igual manera que les han colado la Estrategia Farmacéutica, introducen una Ley con una solución igual de claudicante que contenta a ambos, marcas y genéricos, para no tener que bajar sus precios: un sistema de precios seleccionados de postureo que producirá bajadas testimoniales de precios y una modificación del sistema de precios de referencia, que permitirá mantener artificialmente el diferencial de precios entre ambos, con lo que las marcas podrán superar el precio de referencia sin que se las excluya de la financiación. Y los pacientes y al SNS, que somos los paganos de su orgía de precios, indefensos.
Y si para conseguir sus objetivos tienen que repetir machaconamente una falsedad, pues lo hacen. Confunden o engañan a la sociedad con un descaro que sólo es posible por la seguridad que tienen de controlar perfectamente el discurso y de que los medios afines retransmiten y amplifican sin la más mínima crítica, verificación ni contraste. Así, recientemente el presidente del laboratorio Cinfa (3), líder de ventas de medicamentos genéricos en España, afirmó que “el 50% de los medicamentos genéricos tienen un precio menor de 1,6 euros”. Desde luego no se pueden falsear los datos de manera más escandalosa: de las 1.401 presentaciones diferentes de medicamentos genéricos y biosimilares dispensables en oficina de farmacia que hay en España, sólo 41 tienen un precio inferior a 1,6 euros, en cambio son casi el doble (76) los que tienen un precio de financiación superior a 100 euros. La realidad es que el precio medio de esas 1.401 presentaciones es de 26 euros y la distribución de sus precios por quintiles es la siguiente:
Q1: hay 280 con un rango de precios de <3 euros por envase.
Q2: hay 280 con un rango de precios entre 3 y 7,29 euros por envase.
Q3: hay 280 con un rango de precios entre 7,31 y 15,6 euros por envase.
Q4: hay 280 con un rango de precios entre 15,61 y 36,62 euros por envase.
Q5: hay 280 con un rango de precios entre 36,63 y 507,84 euros por envase.
Si un medicamento que costaba 9,45 euros ha podido bajar su precio hasta costar 2,67 euros y seguir siendo rentable para los 5 laboratorios de genéricos que lo comercializan, ¿cuánto nos están estafando con los 733 tipos de medicamentos genéricos que superan ese precio? ¿Y con los 1.301 que superan los 2,25 euros que costaba el otro medicamento que ha reducido su precio a menos de la mitad y que sigue siendo rentable para otros 8 laboratorios de genéricos?
No, el problema de los genéricos en España no es que sean muy baratos, como repite machaconamente la industria cada vez que tiene ocasión, sino al contrario, que son excesivamente caros. Pero, la verdad ¿a quién le importa?
El Ministerio de Sanidad cumpliendo su compromiso ha elaborado un anteproyecto de ley de Medicamentos y Productos Sanitarios y una vez aprobado por el Gobierno lo ha puesto a información pública antes de elevarlo, para su debate, y complicada aprobación, por el Parlamento.
Sobre el texto que se ha hecho público caben unas primeras observaciones. Es detallado y bastante exhaustivo. Además del texto del anteproyecto de ley, se añaden una gran cantidad de documentos, no sólo los que son de obligada tramitación y consulta ante una norma legal, tanto a instituciones públicas como privadas interesadas; también se añaden otros con información detallada sobre aspectos relacionados directamente con la norma; menos frecuentes de poner a disposición general y que en algunos casos se comentarán, porque son de especial relevancia. Por ello, sólo cabe el reconocimiento al esfuerzo de información y transparencia realizado por parte del Ministerio de Sanidad.
Antes de hacer un análisis necesariamente parcial del contenido del anteproyecto cabe formular algunas preguntas.
¿Es necesario? ¿Es conveniente? ¿Aborda de forma suficiente la problemática del sector? ¿Aporta novedades suficientes para mejorar la prestación farmacéutica y, con ello, la calidad asistencial a la ciudadanía? ¿Va a ser comprensible por esa misma ciudadanía a la que se dirige en primer término? ¿El sector farmacéutico como agente plural, diverso y profundamente asimétrico va a defenderlo, asumirlo y mejorarlo en su tramitación para ayudar a hacerlo realidad?
Intentaré buscar respuestas a algunas de estas preguntas desde el claro compromiso de ayudar a que el proyecto sea una realidad y desde la más absoluta independencia, nacida de la experiencia profesional con diversas partes y agentes del sector al que he dedicado muchas horas y al que no sólo respeto, sino que considero imprescindible para la mejora continua y permanente de una prestación de enorme importancia para la mejora de la calidad de vida de cada persona y, con ello, del conjunto de la sociedad.
Considero de partida que el proyecto es muy necesario.
Haciendo una mirada hacia atrás, siempre conveniente para desde el presente, intentar avanzar de la mejor forma hacia el futuro, cabe mencionar la primera ley del Medicamento de 1990. El gobierno de entonces se había planteado una ordenación actualizada y global del sector del medicamento desde que había llegado al gobierno en 1982. Y se puso a trabajar en ello. Había mucho por hacer. No se disponía de una ordenación mínimamente moderna del sector sanitario. Y por ahí se empezó. Recordar que el 1 de enero de 1986 España se incorpora a la Unión Europea (UE) y en ese mismo año se aprueba la ley General de Sanidad que sigue vigente. Comenzó una nueva era, con una nueva legislatura. Se siguió trabajando por la Dirección General de Farmacia y Productos sanitarios en el proyecto de sacar adelante una ley imprescindible sobre un producto tan básico para la salud pública como el medicamento.
Pasaron cuatro años más, hablando permanentemente con el sector y en coordinación con otros departamentos relacionados con el medicamento, especialmente el de Industria, y es el 20 de diciembre de 1990 cuando se aprueba la primera ley del Medicamento (Ley 25/1990). Constaba de 119 artículos y ocupó 19 páginas del Boletín Oficial del Estado. (BOE). Conviene no olvidar que a lo largo de esos años es cuando se va consolidando el modelo autonómico en nuestro país que no se cierra del todo hasta enero de 2002. Y también debe tenerse muy presente, que en ese nuestro modelo autonómico, la sanidad es objeto de especial atención y son las nuevas administraciones autonómicas las que van a tener muchas competencias sanitarias. Ocho años de intenso trabajo quedaron ahí plasmados.
Hay otras fechas muy importantes en el mundo oficial del medicamento que no se suelen tener muy presentes pero que son de especial trascendencia para entender este proceloso mundo. La Agencia Europea del Medicamento (EMA) no se crea hasta 1995, en Londres, hoy está en Ámsterdam, porque no anduvimos suficientemente atentos y serios para que se estableciera en Barcelona, es la institución máxima responsable de la aprobación, evaluación científica, supervisión y el control de seguridad de los medicamentos desarrollados por empresas farmacéuticas para su uso en la UE.
Hay que esperar hasta el 31 de diciembre de 1997 a que se cree la Agencia Española del Medicamento (AEM) como organismo público con carácter autónomo adscrito al Ministerio de Sanidad y Consumo, en una ley de Acompañamiento lo que tiene su singular significado político.
En esos años se avanza mucho. Se innova mucho. Se descubren y se ponen a disposición de la ciudadanía nuevas y avanzadas terapias y nuevos medicamentos y la UE, junto con las Agencias nacionales y la EMA, modernizan las relaciones entre la industria y los servicios sanitarios en su conjunto, desde los grandes hospitales a las farmacias rurales más distantes.
Los Reglamentos Comunitarios aprobados por la Comisión Europea y las instituciones nacionales avanzan conjuntamente en la normalización del sector, introduciendo modificaciones en la legislación y en el ordenamiento diario. En muchos casos añadiendo complejidad no siempre necesaria y, en otros, ligada a la prestación en sus aspectos técnicos y económicos. La normativa se hace enormemente compleja de entender y de manejar. Se abusa en demasiados casos del Real Decreto-Ley (RDL) aprovechando mayorías parlamentarias estables.
En este continuo quehacer y avanzar se elabora y aprueba el 26 de julio la Ley 29/2006, de garantías y uso racional de los medicamentos y productos sanitarios, que aún hoy mantiene en vigor sus disposiciones finales 2, 3 y 4, por la disposición derogatoria única del Real Decreto Legislativo 1/2015, que consta de 114 artículos, 15 Disposiciones adicionales y 10 Disposiciones transitorias.
Hay que esperar hasta el año 2015 para que se apruebe el citado Real Decreto Legislativo 1/2015, de 24 de julio, por el que se aprueba el texto refundido de la Ley de garantías y uso racional de los medicamentos y productos sanitarios. Este nuevo texto tiene 126 artículos al que acompañan 16 disposiciones adicionales, alguna de ellas de especial relieve como se comentará más adelante. Este texto también fue modificado el 27 de diciembre de 2022, modificación que entró en vigor en junio de 2023 y, por tanto, es en su totalidad el que está en vigor actualmente.
Han pasado ya 10 años y los avances innovadores en todas sus facetas científicas, técnicas, sociales y económicas han seguido siendo muy numerosas y singulares. La UE ha seguido aprobando Reglamentos que son necesariamente incorporables a nuestra legislación. Así que parece absolutamente necesario actualizar y poner al día la regulación estatal de los medicamentos y productos sanitarios.
Se considera no sólo necesario, sino que es conveniente.
Podría optarse como en anteriores ocasiones, por la fórmula del texto refundido que se limita a incorporar ordenadamente las modificaciones obligadas que han tenido que aplicarse para adaptar la norma a la regulación europea o a la impuesta por otros ordenamientos y realidades nacionales. Pero en estos 10 años se han producido muchos cambios sustanciales en el entorno que rodea al mundo del medicamento que aconsejan una reflexión política sobre ellos en el ámbito que le corresponde al Estado, y teniendo muy presente, la internacionalización creciente de la problemática y de las soluciones que se demandan de un sector de alta innovación como es el medicamento y su afectación a la salud pública, como puso tan en evidencia la pandemia del COVID-19 en 2020.
Es importante valorar, dejando al margen intereses corporativos, si el proyecto da suficiente respuesta y de forma adecuada a la actual problemática del sector.
Debe tenerse presente que, aunque estemos ante un instrumento de máximo rango normativo, no puede ni debe entrar en otros ámbitos relacionados con el medicamento, pero que tienen su particular espacio y problemática. No se habla de asuntos de especial incidencia en el sector como son los relacionados con las patentes, ni tampoco del modelo singular de farmacia español o mediterráneo, ni de la permanencia del copago en el sistema. Son importantes problemas que también deben encontrar el momento de debatirse y afrontarse.
Es evidente que el sector del medicamento y de los productos sanitarios es un sector fuertemente intervenido, seguramente uno de los que más, y ello en función de su importancia y afectación a un bien tan superior como es su relación directa, aunque no única, con la salud individual y colectiva. También que por razones complejas a las que en algún aspecto concreto se hará mención, esta intervención es muy asimétrica, con las claras consecuencias que eso conlleva sobre la rentabilidad empresarial.
Se considera globalmente que el anteproyecto aporta novedades suficientes para mejorar la prestación farmacéutica y, con ello, la calidad asistencial a la ciudadanía. Son ordenadamente descritas en la documentación a la que se ha hecho referencia y se sugiere que antes de cualquier valoración general de las mismas se lean con detenimiento. No es un texto refundido como se ha comentado anteriormente.
La ciudadanía ni siquiera de forma minoritaria accede a estos textos, son de compleja comprensión, pero se ha hecho un importante esfuerzo de hacerlos más entendibles en la documentación que lo acompañan y, por ello, los agentes que forman parte del entorno diverso del medicamento sí disponen de elementos para su debate con mucha más información y transparencia que otras ocasiones. Cosa distinta es que se tenga voluntad de reconocer y valorar con rigor todos los espacios que componen la trama del medicamente con claridad, sinceridad y voluntad globalizadora.
El debate está servido y ahora cabe preguntarse si el sector farmacéutico como agente plural, diverso y profundamente asimétrico va a defenderlo, asumirlo y mejorarlo en su tramitación para ayudar a hacerlo realidad.
Las primeras reacciones que se están escuchando son, como era previsible, de crítica generalizada y diversa. Cuando se tiene la firme determinación de introducir cambios en un sistema complejo y estos son novedosos, y con mayor o menor incidencia en riesgos y beneficios, afectando prácticamente a todos los subsectores que lo integran en mayor o menor medida, cabe esperar de los afectados esa reacción negativa ante el posible cambio antes de hacer una valoración reflexiva y de conjunto.
En el entorno mediático en que estamos inmersos, también parece inevitable que se planteen críticas y comentarios, que al margen de su interés más o menos legítimo, sean puramente demagógicas e incluso falsas.
Estos planteamientos no aportan nada al debate que una norma de esta importancia merece.
Se ha dicho y repetido con rapidez, que la norma contempla y regula de nuevo las subastas de medicamentos como instrumento de bajada de precios. El anteproyecto introduce una nueva figura de “precios seleccionados” que explica con mucho detalle en documentos adjuntos y que nada tiene que ver con una subasta, que se centra en un proveedor único que ofrece el precio más bajo y excluye a los demás. El sistema de precios seleccionados es todo lo contrario. Permite la competencia, con transparencia, y añade un elemento de diversificación que no deja fuera de la financiación pública a ninguno de los medicamentos que quedan fuera de la horquilla que recoge y ordena esos medicamentos seleccionados. Las “marcas” y otros medicamentos excluidos de la subasta quedaban en una situación discriminada. Ahora no; los que quedan fuera siguen pudiendo ser elegidos por los ciudadanos, que aportan la diferencia entre el precio más bajo financiado y el que esos fabricantes o proveedores mantienen. Por supuesto que es una medida novedosa y atrevida, pero es en primer lugar, valiente, porque todo lo que sea fomentar un sano ejercicio de libertad y pluralidad en un sector con tantos recovecos, ayuda a una justa y mayor redistribución y, de forma colateral, a fomentar la presencia de más agentes compitiendo y, con ello, menos riesgo de falsos monopolios a la vez que modera el riesgo de desabastecimiento. Esta medida debería en vez de criticarse sin argumentos o con comparaciones falsas, tomarse como una decisión inteligente que se repite en otras de las reformas propuestas que también se comentarán.
En relación con la gran asimetría del sector cabe hacer una reflexión. En primer lugar, es una realidad que en el subsector de la producción están presentes grandes compañías multinacionales, que son pocas, porque han ido creciendo a través de importantes fusiones, con un conjunto mucha más numeroso de empresas medianas y pequeñas. En cada subsector de estas compañías vuelve la diversidad. Algunas están y fabrican en Espala y otras no, e incluso fabrican producto intermedio o terminado que luego redistribuyen por otros mercados. Solo algunas de estas grandes compañías hacen investigación real. Otras que la hacían han centralizado la investigación en otros lugares. Algunas colaboran con centros de investigación, otras hacen y colaboran en ensayos clínicos, que siendo una importante tarea dista mucho de la I+D de mayor trascendencia. Otras mantienen relaciones con universidades o start-ups investigadoras muy focalizadas, que una vez cerca de un nuevo producto y sin suficientes medios para llevarlo a su fin, lo hacen suyo beneficiándose de lo que significa un nuevo medicamento innovador a través de la patente. Otras se centran en los genéricos y otras, pocas, en los biosimilares, y finalmente, otras fabrican medicamentos sin patentes, no genéricos y que en algunos casos les han “vendido” compañías más grandes que han dejado de tener interés por medicamentos más añejos pero que siguen teniendo interés terapéutico, pero que por su bajo precio dejan de serles atractivos. Por supuesto parece evidente que unas y otras nos pueden valorar las reformas que el proyecto contempla con la misma mirada y con las mismas reservas, pero para todas, el proyecto quiere señalar un nuevo camino de mayor transparencia y competencia leal, dentro de la diversidad y de la complejidad. Y eso es también destacable e inteligente.
Lo que es singular en el sector productor, es qué teniendo una problemática diferente y en algunos casos distante, una gran parte de estas empresas se encuentran juntas en una gran “patronal”, Farmaindustria, interlocutor muy destacado de la administración sanitaria y cuya opinión en forma de propuestas al texto, será intenso, extenso y que ofrecerá también una ocasión de estudio y reflexión. Lo que puede adelantarse con poco riesgo de error, es que será crítica con el proyecto, especialmente en todo lo que se refiere a las aportaciones del porcentaje sobre ventas que el proyecto contempla en distintos apartados y especialmente en la Disposición Adicional 6ª.
En este sentido el anteproyecto ha optado por mantener un conjunto de disposiciones relacionadas fundamentalmente con descuentos selectivos que se han ido produciendo a lo largo de los años en forma de RDL y Reales Decretos (RD). Normas del año 2000 y singularmente de los años 2008,2009 y 2010.
Es preciso tener presente y recordar que mientras en el conjunto de la distribución de competencias a lo largo del proceso de consolidación de las Comunidades Autónomas estás tienen un amplio e importante marco competencial en el mundo del medicamento que nos ocupa, esta sigue siendo una competencia exclusiva del Estado en que colaboran activamente las Comunidades.
El principal objetivo de los RDL y RD a que se ha hecho referencia era la disminución del gasto farmacéutico, bien porque su evolución superaba una determinada racionalidad presupuestaria, o por la coincidencia con crisis económicas coyunturales. Esta realidad merece un paréntesis aparte para volver luego a estas normas y su permanencia. Estas actuaciones también han sido compatibles con otras de incentivación selectiva para determinadas acciones que se consideraban o prioritarias o necesarias para cubrir suficientemente determinadas demandas sanitarias.
Lo que ha sido una constante en la mayor parte de estas medidas ha sido su singularidad o urgencia. Pero lo que se ha constatado es qué con excesiva frecuencia, aquello que tenía una clara razón coyuntural, se ha consolidado en vez de reformar la norma para mejorar su coherencia cuando la razón y el objetivo de la medida urgente hubiera dejado de existir. Ha parecido más útil y fácil mantener la medida coyuntural que adecuar el conjunto, y el sector con su capacidad de adaptación y con una reserva conservadora ante los cambios estructurales, ha preferido irse adaptando.
En parte, este proyecto plantea medidas nuevas estructurales de racionalidad y de control del gasto a la vez que mantiene aquellas otras medidas coyunturales relacionadas también con el gasto y, sobre todo, con los ingresos. No es aventurado suponer que será criticado por lo nuevo, y no incentivado, a suprimir lo coyuntural.
En este sentido pudiera ser también valiente y positivo para dar más pasos adelante en una senda de racionalidad, modernidad y eficiencia suprimir normativa coyuntural antigua y sustituirla por medidas estructurales de forma que el objetivo final del gasto fuera el que la nueva norma contemple. En este sentido se adelanta que las cifras que se aportan de disminución del gasto, superiores a los 1.300 M€, se consideran demasiado optimistas e incluso ni siquiera eficientes.
También se considera que los descuentos contemplados en los RD 8 y RDL 9 que afectan a toda la cadena del medicamento y con proporcionalidad en función de las ventas, conllevan una importante burocracia para el sistema, lo que afecta a la efectividad y a la productividad de los responsables, tanto del sector como de las administraciones. Por ello, se propone que debiera estudiarse su derogación y ser sustituidos por una disminución del margen farmacéutico. Para las farmacias de menores ventas, que deben ser protegidas en el ámbito rural de poca población y con tendencias de despoblación, al final de año con el dato objetivo de ventas, este podría ser completado en la cantidad adecuada, siempre sobre el margen general establecido, y puede ser el momento para añadir una atención singular a estas farmacias tan necesarias y contemplar el pago de acuerdo con las Comunidades Autónomas, de unas determinadas guardias que reconozcan su especial dedicación y disponibilidad que completan la capilaridad asistencial.
Siguiendo con la vigencia de los RDL y RD citados y su importancia en el gasto, se comparte en líneas generales el planeamiento de la Disposición Adicional 6ª, también longeva, pero que contempla la extensión de su aplicación al gasto derivado de las ventas hospitalarias. Por supuesto va a encontrar el rechazo por parte de Farmaindustria en este caso en el papel de defensora de las grandes compañías que son las que disponen de una mayor parte de los medicamentos con patente, generalmente más nuevos y de más alto precio y de venta mayoritaria a los hospitales. Pero es la ocasión de poder dejar claro que estas importantes compañías tienen la parte más sustancial del conjunto del sector. Los datos son indiscutibles en porcentaje de ventas y de beneficios. No se puede, ni se debe tener todo, incluyendo esos no siempre justificables privilegios de confidencialidad encubierta en el manto protector de la patente y que se ponen más evidencia precisamente en las ventas a los hospitales.
También es el lugar en el proyecto en el que se hace referencia al plan Profarma que lidera el Ministerio de Industria, energía y turismo, qué en sentido contrario a la aportación porcentual en ventas al sistema, determinadas empresas reciben unas bonificaciones en función de una serie de condicionantes y comportamientos evaluables y cuantificables. Se sugiere darle también una vuelta. Ha cambiado mucho desde aquellos lejanos años 80 en que fue puesto en marcha en un entorno de entrada en Europa y como tantas otras cosas que se han comentado, ha evolucionado más lento en su visión de una pequeña parte del sector que del conjunto del sistema por aquella tendencia inercial de todo lo que tiene una fuerte composición administrativa. Ni están todas las empresas que son, ni son todas las que están en sintonía con las orientaciones generales que inspiran el proyecto.
Los RD citados no sólo tienen afectación a los directamente dirigidos, empresas y farmacias, y en una muy pequeña parte a otro de los agentes del sector, como es la distribución, que es generalmente poco protagonista en las normas y con frecuencia demasiado en la vida real del colectivo. En la complejidad de gestión administrativa a la que se ha hecho referencia por la aplicación de estos decretos, no todos los agentes participan con la misma proporcionalidad y transparencia. Téngase presente como un argumento más para tomar en consideración su supresión, que aportaría sencillez y transparencia y evitaría complejidades no siempre justificadas y comportamientos fronterizos.
En resumen, prescindir de estas normas de hace años y coyunturales puede mejorar el conjunto de la gestión del sistema, hacerlo más transparente y las cuentas pueden perfectamente seguir saliendo y distribuir sus efectos con pequeñas diferencias a los distintos agentes que, por supuesto, van a levantar la voz, con menos razón que volumen, si se hacen bien los cálculos y se habla con franqueza.
Puede ser el momento de pasar de las letras a los números del gasto público para focalizar mejor la realidad del sector.
Se observa de forma constante un incremento del gasto farmacéutico en su conjunto en un porcentaje que sigue dentro de un margen razonable al resto de la evolución de crecimiento de la economía, de la población, del envejecimiento de la misma, del incremento innovador exponencial que se adentra en esferas biológicas y bioquímicas impensables hace muy pocos años, en la prevención y en los indicadores de salud pública.
Este crecimiento se hace más intenso en el gasto hospitalario que prácticamente iguala en precio de laboratorio al de farmacia. Alrededor de 13.000 M€ cada uno. Menos de un millar de hospitales y más de 22.200 oficinas de farmacia, que dan trabajo a más de 80.000 empleados, de los que más de 30.000 son farmacéuticos, y que siguen creciendo en determinadas ciudades y disminuyendo como se ha indicado anteriormente con preocupación en el mundo rural aquejado de despoblación. Ese gasto lo prescriben 38.000 médicos de atención primaria que de media prescriben en un año más de 300.000 €, cantidad entre cinco y seis veces su salario. Y 93.000 médicos de hospital acompañados de algo más de 3.000 en periodo de especialización (MIR). No todos prescriben, pero puede estimarse una media de prescripción de aproximadamente 200.000 €.
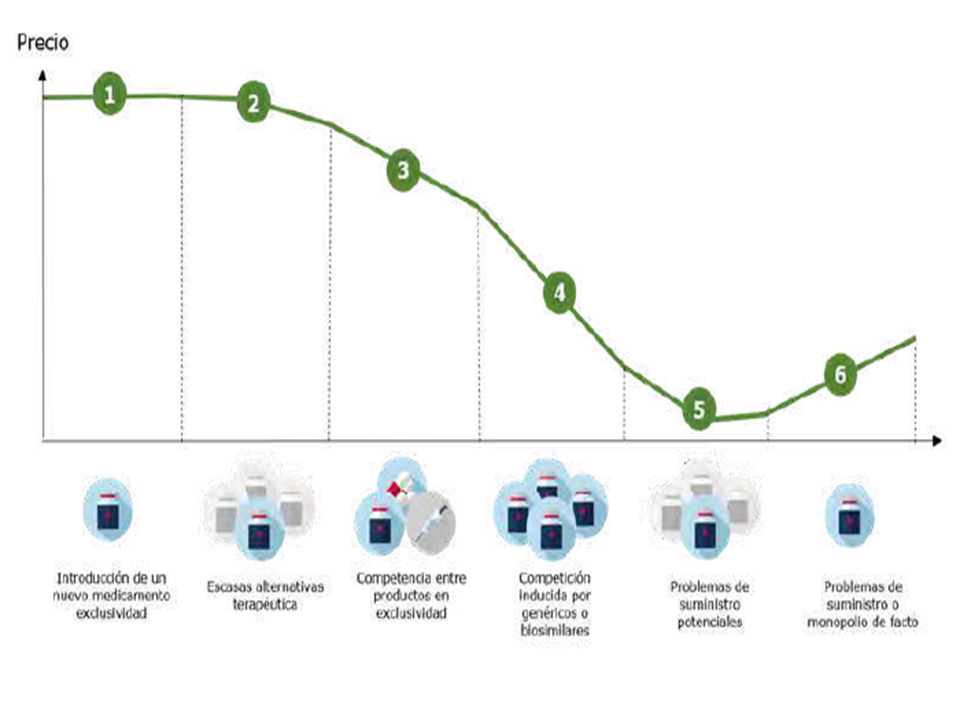
El porqué de añadir estos números y ahora es por la gran información económica que acompaña al proyecto de ley y sobre todo para intentar relacionarlo con el muy interesante gráfico que se recoge en la página 6 del documento ejecutivo elaborado por el Ministerio y que publica en su web simultáneamente al proyecto.
En este grafico se expresa de una forma muy novedosa y que refleja gráficamente mejor que con muchas palabras la realidad y ciclo temporal de los medicamentos. De todos. Desde los más innovadores con patente que ocupan el tramo 1, a los que al poco tiempo de esa vida real 3 o 4 años por diversas circunstancias tienen pocas alternativas terapéuticas, y con ello poca competencia y ligera bajada del precio, lo que se representa en el tramo 2. Avanza el tiempo y se incrementa la competencia frente a la exclusividad, estamos en el tramo 3 y lógicamente baja el precio medio, que se intensifica con una competición claramente inducida por los genéricos y biosimilares y que refleja el tramo 4. Es interesante fijarnos en el tramo 5. Intenta reflejar ese conjunto de medicamentos muchas veces sin patente y que engloba a medicamentos esenciales y básicos, que es preciso, no sólo proteger, sino estimular y defender porque están potencialmente más expuestos a la disponibilidad, acrecentada porque su precio es bajo; finalmente el tramo 6 del gráfico, se refiere a medicamentos antiguos, con marca, y sin posibilidad de competencia genérica. y que en muchas ocasiones son un monopolio de hecho y que también son de un uso frecuente y necesario. El gráfico intenta que las áreas bajo la curva representan el gasto parcial de cada tipo. Es un claro reflejo de la realidad. Y sobre ella el proyecto propone medidas que cuantifica. Con el sistema de precios seleccionados conseguiría bajar la curva en los tramos 2 y 3 en una cifra que se cuantifica en 800 M€. También se propone que la curva en el tramo 5 suba para proteger vía precio esos medicamentos esenciales y cuantifica el esfuerzo en 150 M€.
Nunca hasta ahora se había expresado con tanta sencillez la realidad del tan poblado número de medicamentos que hemos construido y que supera las 16.000 presentaciones.
Es preciso también hacer una referencia a otros importantes agentes del sector. Ciertamente Farmaindustria es un interlocutor de especial relevancia por su envergadura, su solidez, su profesionalidad, su experiencia y su internacionalización, pero también es preciso hacer referencia a la Asociación Española de Medicamentos Genéricos (AESEG) y a la Asociación Española de Biosimilares (BioSin), las patronales de genéricos y biosimilares. Seguramente serán los interlocutores que muestren más reticencias a la innovación comentada de los precios seleccionados, que pueden ver como una amenaza a ese término tan acuñado: ”claw back”, ese retroceso a todos los efectos. Esos descuentos prolijos y que pretenden generar una competencia relacionada fundamentalmente con el margen de la farmacia, sin retorno alguno para el ciudadano, ni para la administración, que participa como mucho sí la transparencia es la adecuada, en un determinado ingreso fiscal. La implantación del sistema de precios seleccionados ayudará a simplificar la gestión de la oficina de farmacia que a este nivel de gestión es la pieza más débil del conjunto del sistema. Seguramente tampoco recibirán con entusiasmo la mayor libertad del paciente de poder elegir medicamentos fuera de la horquilla de precios seleccionados pagando sólo la diferencia a la que se hizo referencia en aquel apartado. Siempre ha sido valorar y ponerle precio a la libertad.
Se considera preciso hacer una referencia a otros representantes colectivos de sectores importantes del sector. Los colegios profesionales y las patronales farmacéuticas.
La oficina de farmacia tiene una doble personalidad. Estableciendo privado con una función pública sanitaria de importancia significativa a la que une un componente de pequeña y mediana empresa, lo que no se quiere reconocer ni siquiera fiscalmente.
Dado el elevado número de oficinas de farmacia y su presencia en todo el territorio, su organización corporativa sigue fundamentalmente la organización provincial que se instauró en el siglo XIX. Así se configuran los Colegios Provinciales de colegiación obligatoria para todos los que están en las oficinas. Lógicamente han tenido también que configurarse a partir del final de la estructuración autonómicas, estructuras colegiales regionales, en las Autonomías pluriprovinciales. Todo el entramado territorial tiene su máxima integración en el Consejo Superior de ámbito estatal que coordina la tarea de los colegios provinciales y tiene la máxima representación institucional del colectivo ante determinados interlocutores y tareas y cuyos máximos representantes son elegidos por estos. Las competencias de los colegios profesionales se focalizan en la defensa de los derechos de sus afiliados y de forma muy destacada, en la preservación de los valores relacionados con las competencias profesionales y con la vigilancia del desempeño de sus tareas, vigilando la ética profesional y velando también por la debida formación de sus integrantes.
Es bien conocido que en el caso que nos ocupa las funciones de hecho de los colegios farmacéuticos son singulares al tener un papel de intermediación con los responsables institucionales del gasto farmacéutico. Esta singularidad configura una estructura y comportamiento y realidad económica también única. Al margen del respeto a cualquier opinión, es de difícil justificación que un gasto público perfectamente regulado y cuya gestión y responsabilidad recae en las Comunidades Autónomas, como cualquier otro gasto público, se gestione a través de un intermediario corporativo. Es hacer más corporativo a un órgano corporativo. Por supuesto esta intermediación conlleva un gasto que es el que mantiene la estructura corporativa y que se detrae del beneficio de la oficina de farmacia. A nadie se le escapa que esa gestión del pago del gasto farmacéutico no se realiza directamente por los órganos colegiales, sino por empresas contratadas, que también añaden su coste al proceso. Eso configura al colegio, añadiendo a sus funciones genuinas, unas funciones prácticamente de interlocutor privilegiado con la administración pública en tareas no sólo profesionales, sino también de ordenación en la tarea de las oficinas de farmacia y afecta a su componente empresarial que hace que prácticamente no tengan presencia significativa las instituciones patronales del sector de oficinas de farmacia, a las que prácticamente quieren sustituir, lo que aumenta la debilidad de éstas como interlocutoras, frente a las potentes patronales del sector de empresas farmacéuticas.
Debería, por tanto, en aras de modernizar y poner en su lugar competencial a cada uno, revisarse esa función de intermediación económica de los colegios farmacéuticos, lo que afectaría también, al Consejo General, ya que al modificarse la de los Colegios provinciales precisaría una revisión profunda en su configuración democrática y representativa. Se puede orientar y estimular desde el gobierno esa acción modernizadora a la vez que se desarrolla el contenido de esta ley, que tiene como claro objetivo la mejora y actualización del conjunto del sector.
El proyecto contempla diversificar la responsabilidad de prescripción a profesionales de la enfermería y la fisioterapia. Se mantiene el previo diagnóstico por el personal médico. La prescripción en la que se centrarán es presumible que se dirija a los productos sanitarios, con lo cual se mejorará la disponibilidad de estos. También se avanza en la lógica mayor responsabilidad de los farmacéuticos a la hora de sustituir un mismo medicamento por otra de sus formas de presentación sin necesidad de volver a la intervención del médico e incluso en circunstancias especiales por medicamentos similares. Pequeños pasos en el camino de la racionalidad frente al rígido corporativismo y pensando en el paciente como principal destinatario.
Como conclusión hay que indicar que estamos ante un importante intento de modernización de un sector de especial relevancia pública, ligada a un objetivo del estado de bienestar al formar parte el sector del medicamento del componente básico de preservación, prevención y mejora de la salud individual y colectiva. El esfuerzo de los impulsores y redactores del proyecto merecen un claro reconocimiento, no han escogido el camino más fácil de la continuidad de los textos refundidos, sino de ahondar en las complejidades de un sector plural, diverso, de alta cualificación e internacionalización. Queda camino por recorrer en estos momentos claves que deben dirigirse a una mayor unidad europea que no sea meramente formal, sino real y ejecutiva, política, económica y cultural, para que el medicamento también sea un elemento de referencia, de uso, de consumo de accesibilidad igual para todos. Este proyecto no da por acabado el camino, pero da unos pasos decididos, valientes, claros e inteligentes en esa dirección.
De nuevo, un breve artículo de Dean Baker, donde expone una alternativa enormemente sugerente a las políticas arancelarias de Trump: “no respetar los monopolios estadounidenses de patentes y derechos de autor”. Una posibilidad que en un hipotético caso de aplicación de esta medida supondría un cambio en la percepción mundial de las patentes. Pero además supondría un ahorro muy considerable para los sistemas de salud y para las y los ciudadanos. Sin monopolios de patentes, concluye Baker, se produciría una diferencia en el gasto que, ascendería a unos 5.000 dólares para cada hogar del país.
La mayoría de las represalias que los países planean tomar en respuesta al aluvión de aranceles de Donald Trump incluyen aranceles más altos y restricciones a las importaciones. Estas medidas pueden perjudicar la economía estadounidense, pero también perjudicarán al país que las impone. La lógica es que las medidas se diseñarán de forma que el sufrimiento en Estados Unidos sea mayor que el que experimente el otro país.
Es probable que esto sea cierto, pero la UE, Canadá y otros nuevos enemigos pueden ir un paso más allá. Pueden implementar medidas de represalia que perjudicarán gravemente a Estados Unidos, a la vez que benefician a sus propias economías. En concreto, pueden anunciar una política de no respetar los monopolios estadounidenses mientras Donald Trump siga con su absurdo juego arancelario.
Hay mucho dinero en juego. El año pasado, Estados Unidos recibió casi 150.000 millones de dólares en regalías y derechos de licencia. Eso representa más del 5 % de todas las ganancias corporativas después de impuestos.
Y esto se refiere solo a los derechos de autor. No incluye todos los casos en los que la propiedad intelectual está incorporada al producto. Por ejemplo, no se contabilizaría el valor del software de una computadora estadounidense enviada a Canadá o la UE. Los fabricantes estadounidenses de computadoras tendrían muchas más dificultades para competir en el extranjero si sus competidores pudieran usar el sistema operativo Windows y otro software de Microsoft sin coste alguno.
De hecho, existen precedentes de no respetar las patentes de países en conflicto. Estados Unidos utilizó la Ley de Comercio con el Enemigo durante la Primera Guerra Mundial para permitir la concesión de licencias obligatorias de patentes pertenecientes a empresas o ciudadanos alemanes. Esto significaba que las empresas podían usar estas patentes sin el permiso de los titulares alemanes, siempre que pagaran una modesta tarifa de licencia establecida por el Gobierno estadounidense. Canadá, la UE y otros socios comerciales de EE. UU. pueden seguir el mismo camino.
Lo interesante de esta forma de represalia es que, en realidad, beneficiaría directamente a los consumidores de los países que optan por esta vía. Por ejemplo, podrían obtener los medicamentos más recientes para tratar el cáncer o las enfermedades cardíacas como genéricos baratos, en lugar de pagar los miles o decenas de miles de dólares que podrían cobrar Pfizer o Merck.
Nadie les impediría hacer copias de las últimas películas de Disney y distribuirlas libremente. Y las computadoras serían mucho más baratas si no tuvieran que pagar licencias a Microsoft por su software. Este sería un gran ejemplo de cómo hacer el bien haciendo el bien.
Supongo que, si nuestros socios comerciales optaran por esta vía del libre comercio como represalia, también provocaría serios temores en las empresas estadounidenses de una forma que otras formas de represalia jamás podrían. Si la gente de otros países se acostumbrara a medicamentos y computadoras baratos, y a versiones gratuitas de las últimas películas de Hollywood, podría ser difícil volver al viejo sistema. La fábrica de dinero para muchas grandes empresas estadounidenses podría verse descarrilada para siempre.
La mayoría de nosotros aquí también estaríamos muy contentos con un mercado más libre de esta manera. Si no tuviéramos monopolios de patentes sobre medicamentos con receta, probablemente estaríamos gastando cerca de 100.000 millones de dólares al año en lugar de 650.000 millones. La diferencia asciende a unos 5.000 dólares por cada hogar del país.
El verdadero libre comercio, no las falsedades que incluyen en nuestros acuerdos comerciales, sí ofrece grandes beneficios. Los ricos amigos de Trump en Mar-a-Lago estarían muy descontentos si la gente de otras partes del mundo, y posiblemente también aquí, se dieran cuenta de cuánto podrían beneficiarse eliminando o debilitando los monopolios de patentes y derechos de autor.
Sería un gran acto de valentía por parte de los líderes extranjeros optar por el libre comercio para combatir el disparatado régimen arancelario de Donald Trump, pero probablemente sería muy eficaz para poner fin a su absurda maniobra. Ya veremos si empuñan la espada.
Interesante, descriptivo y claro artículo de Salud por Derecho donde, entre otros temas, se abordan algunas de las propuestas relacionadas con el Tratado de Pandemias de la Organización Mundial de la Salud, relacionadas con la transferencia de tecnología y el sistema de acceso y reparto de beneficios.
La Unión Europea ha adoptado una postura crítica frente a algunas de estas propuestas, defendiendo mecanismos voluntarios para que la industria comparta tecnología y conocimiento durante las emergencias sanitarias, y oponiéndose a compromisos obligatorios que garanticen el acceso rápido a ese conocimiento y permitan ampliar la capacidad de producción de productos farmacéuticos.
En este análisis explicamos cómo esta postura es incoherente con otras posiciones y compromisos asumidos por la UE, donde ha apoyado medidas obligatorias y cómo tampoco se corresponde con los recursos destinados y el papel de los Estados de la Unión Europea en la innovación y desarrollo (I+D) de productos farmacéuticos para pandemias. Defendemos que la UE podría beneficiarse de un mecanismo que incorporara medidas obligatorias para la transferencia de tecnología y el reparto de productos farmacéuticos en futuras pandemias.
1. Europa ya apoya principios similares de no voluntariedad en otros foros
La discusión sobre si las medidas para favorecer la transferencia de tecnología durante una pandemia deben ser voluntarias u obligatorias ha sido un punto de tensión durante las negociaciones. La experiencia de la COVID-19 demostró que confiar exclusivamente en medidas voluntarias es ineficaz, Un ejemplo de ello es la falta de contribuciones a mecanismos como C-TAP. Expandir rápidamente la capacidad de producción a través de la transferencia tecnológica durante una emergencia es esencial para proteger la salud global. Esta expansión requiere una combinación de medidas voluntarias y obligatorias, como señala el Global Health Centre.
La falta de mecanismos ágiles para expandir la capacidad de producción de vacunas equivale a pérdidas de vidas y un aumento innecesario de los costos sanitarios. Existen medidas obligatorias, como las licencias obligatorias, que están amparadas por el derecho internacional (Acuerdo ADPIC) y han sido utilizadas por países de todos los niveles de ingresos, incluidos Estados miembros de la UE. Por ejemplo, Alemania y Hungría modificaron su legislación durante la pandemia para agilizar el otorgamiento de estas licencias.
La UE es plenamente consciente de la necesidad de este tipo de medidas en contextos de emergencia. Mientras en Ginebra se opone a incluirlas en el Tratado de Pandemias, en Bruselas se está negociando precisamente lo contrario, la obligatoriedad de compartir secretos comerciales para la aplicación efectiva de licencias obligatorias en la UE.
Uso de cookies
Este sitio web utiliza cookies para que usted tenga la mejor experiencia de usuario. Más info