ORIGINAL. Revista n.º 48 de Mayo 2026

Colectivos en Lucha por la Sanidad Pública es un movimiento estatal de reciente creación, donde se han unido distintas plataformas y organizaciones de las CCAA.
Al inicio de su actividad ha organizado dos actos en el contexto del cumplimiento de los 40 años de la Ley General de Sanidad donde plantear: la Participación Comunitaria y el blindaje Constitucional de la Sanidad Pública y Universal, como derechos fundamentales.
La Ley General de Sanidad con sus luces, como, la implantación de la universalidad, la financiación vía impuestos, la integración y cohesión de las estructuras sanitarias de distinta titularidad pública bajo un solo sistema sanitario y con sus sombras, con los articulos 66, 67 y 90, de la LGS que perpetúan la coexistencia del sistema sanitario privado, así como la persistencia de un sistema sanitario distinto para 2 millones de habitantes.
En el primer acto se abordaron temas tan importantes como los inicios de la reforma de la Atención Primaria, con la fuerza y la ilusión de aquellos momentos, las dificultades encontradas y los tiempos en que se generalizó su implantación. Y como este proceso fue paralizado por la corriente neoliberal y su traducción en el Informe Abril, al mismo tiempo que se iniciaba la promoción del consumo sanitario, que Atención Primaria no pudo contrarrestar por voluntad política. Y el resultado de esta dinámica fue la Ley 15/97 de Aznar, apoyada por todos los grupos políticos de entonces; las únicas excepciones en rechazarla y votar en contra fueron Ángeles Maestro de Izquierda Unida (I.U.), y el Bloque Nacionalista Gallego (B.N.G.). Esta ley supuso abrir la puerta totalmente a la privatización del Sistema Sanitario Público para que se pudiera llevar a cabo con más facilidad.
Es con la crisis del año 2008 cuando la Atención Primaria es deteriorada con más fuerza, llegando a una temporalidad entre sus profesionales de un 23%. El grueso de los más de 13.000 puestos de trabajos directos destruidos entonces eran profesionales sanitarios de Atención Primaria. Este deterioro se profundizará aún más con el Real Decreto Ley 16/2012- Con su aplicación el Sistema Nacional de Salud sufre un paso aún más importante en su debilitamiento y entrega a las empresas privadas. Se inician los copagos antes inexistentes y se pierde la Universalidad.
Con el RD se estaba preparando y facilitando un producto más atractivo para las empresas privadas.
En la fase de recuperación de la crisis, la dotación de la Atención Primaria no experimenta mejora alguna; estaba claro que con una estructura deteriorada la privatización es más rentable y así habrá más pacientes que vayan directamente a los especialistas y a las estructuras hospitalarias privadas que es donde se sitúa el negocio más beneficioso.
Este análisis de los rasgos fundamentales de la evolución del Sistema Sanitario Público fue ampliado y reforzado con la exposición de Audita Sanidad, con su trabajo: Estrategia
neoliberal y Privatización de la Sanidad Pública. Su descripción de la privatización hospitalaria en la Comunidad de Madrid, con la utilización los modelos PFI y PPP, así como la de la corrupción que se evidencia una y otra vez en la estrategia de convocatoria y resolución, de contratos menores de la Comunidad de Madrid demuestra una y otra vez el proceso de desmantelamiento del sistema público sanitario
El segundo acto organizado, se realizó el día 22 de mayo en el Congreso de los Diputados. En el mismo se expuso en primer término, como se llevó a cabo la desprivatización del llamado “modelo Alzira” buque insignia de la privatización sanitaria, por el que se había entregado a la empresa Ribera Salud en la C. Valenciana la gestión del hospital así como posteriormente de la Atención Primaria del Área mediante una concesión administrativa.
Esta reversión y recuperación de la gestió pública directa fue realizada de forma impecable gracias a la voluntad política, no sin grandes dificultades provocadas por demandas judiciales y presiones mediáticas y de todo tipo por la empresa Ribera Salud. Durante el proceso de reversión se pudieron comprobar de forma evidente todo tipo de carencias a nivel de recursos humanos y materiales. La lógica de negocio de Ribera Salud estaba por encima de la salud de los ciudadanos y ciudadanas y de los profesionales sanitarios, mientras menos recursos más negocio. Esta desprivatización y como se llevó a término con éxito es un ejemplo de que si se puede realizar, pese a todas las dificultades y que supone un claro mensaje: el negocio debe estar siempre fuera de todo proceso en relación con la salud de la población.
Posteriormente las Asociaciones de Vecinos representadas expusieron la necesidad mejorar el sistema sanitario público y la exigencia a a los representantes parlamentarios de responder a las demandas de la población, defendiendo el sistema público. Al mismo tiempo, necesidad de extender la conciencia en la ciudadanía de la exigencia de un sistema sanitario público, que responda con igualdad y solidaridad para todas y todos. Y como la participación debe realizarse como ciudadanía, no sesgada con otros perfiles
Las características de este proceso implican objetivos definidos y más participacion ejecutiva.
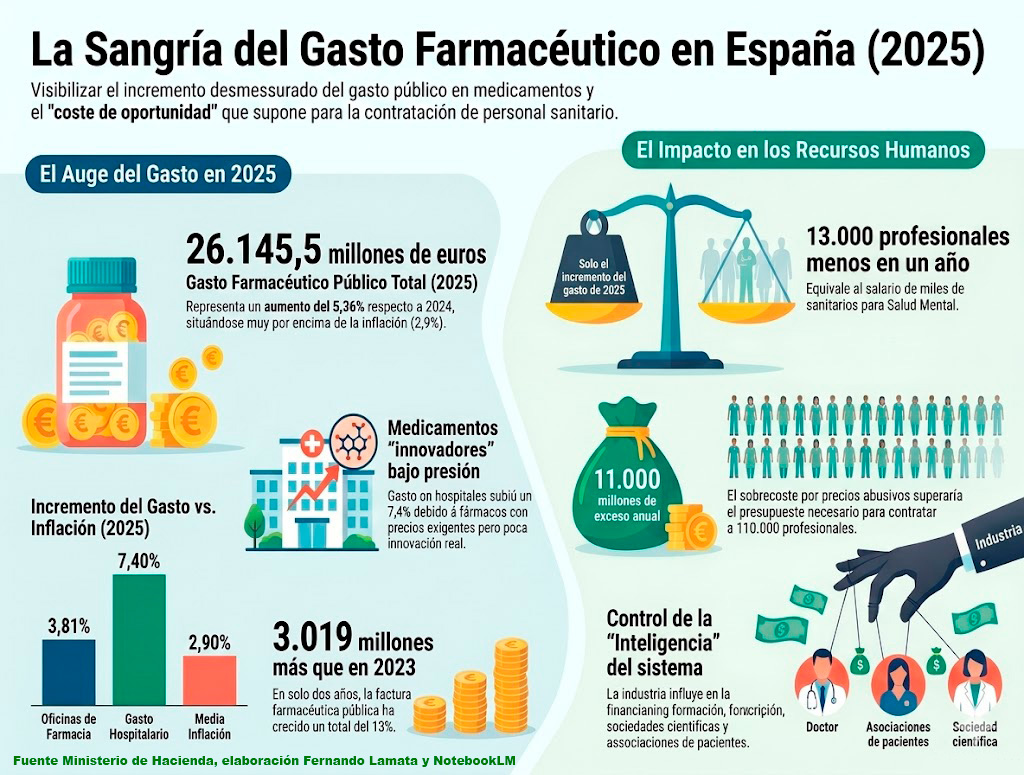
Finalmente, la Asociación para un Acceso Justo al Medicamento AAJM expuso la importancia notable que el gasto farmacéutico tiene para el Sistema Nacional de Salud y el riesgo, que supone el incremento constante y notable del gasto farmacéutico en España, fundamentalmente el gasto hospitalario para su supervivencia . El incremento del gasto farmacéutico en el 2025 permitiría por ejemplo contratar a 13.000 profesionales.
Este gasto descontrolado se debe a los precios escandalosos que las grandes empresas farmacéuticas imponen a los denominados “medicamentos innovadores” término con el que se enmascara la realidad de medicamentos de marca sometidos a un monopolio durante periodos de exclusividad, muy prolongados, en muchas ocasiones más de 20 años. Estos precios tan elevados que no corresponden con los costes reales de investigación y producción se debe a un monopolio que permite a la BigPharma fijar el precio más alto posible sin ningún tipo de límite y sin la obligación de explicar sus costes reales. Los fármacos de altísimos precios en realidad tienen costes muy bajos.
En España el proceso final de financiación pública y fijación de precio, para un medicamento determinado, lo realiza la Comisión Interministerial de Precios de los Medicamentos cuyo margen de negociación es escaso, dado que el precio de referencia es el establecido de manera unilateral por las empresas farmacéuticas en Estados Unidos.
Los sistemas sanitarios públicos en Europa y en España están fuertemente sometidos a las presiones de los lobbies de la industria farmacéutica que abarca todo el ámbito de la política farmacéutica. Actúan sobre los decisores políticos y sobre los profesionales médicos cuya formación continuada depende en la mayoría de las ocasiones de la financiación de la industria farmacéutica. Su área de influencia a través de la financiación incluye a pacientes y sociedades científicas.
En el intervalo entre las dos mesas, tuvimos la oportunidad de escuchar la intervención de una farmacéutica que explicó el papel del laboratorio israelí Teva en el genocidio realizado por Israel al pueblo palestino en Gaza y su papel directo a través de un entramado empresarial. En su intervención llamó al boicot a los productos del mencionado de laboratorio, tanto a nivel de profesionales farmacéuticos y de comunidades autónomas como de los propios ciudadanos y ciudadanas
Colectivos en lucha por la sanidad pública, ha comenzado a caminar. Confiamos profundamente en una ciudadanía comprometida en la defensa de nuestro sistema sanitario público. Consideramos que es clave desarrollar un papel activo para conseguir, recuperar y mejorar un sistema público imprescindible para la salud.
Sin duda se puede.
Amparo Botejara
Colectivos en Lucha por la Sanidad Pública
- En las Jornadas “40 años de la Ley General de Sanidad y la Participación Comunitaria hoy” asistieron como ponentes: Pedro Sabando, Asunción Prieto Orzanco, Cristina de la Cámara, Vicente Losada, Amparo Botejara, Juán Gervás y Juan Antonio Caballero, en la 1ª (24 de abril/26), y en la 2ª (22 de mayo/26) Carmen Montón, Isabel González Rodríguez, Remigio Cordero, Ramón Gálvez, Diego López Garrido, Ana Rosa Encinas y Julio González.
Ver Jornadas completas en los siguientes links: 1ª. Jornada. / 2ª. Jornada.