Presidente de la Asociación Acceso Justo al Medicamento (AAJM).
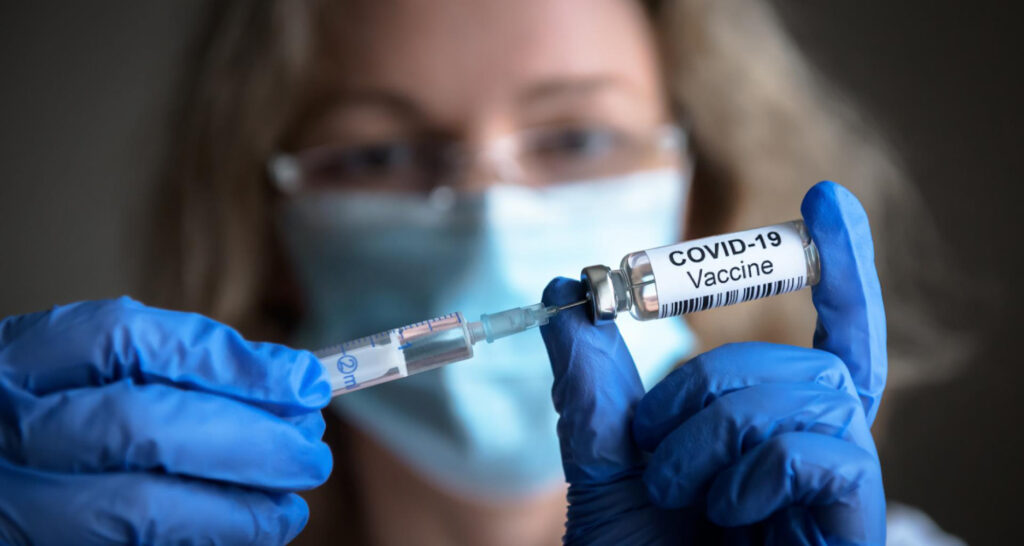
Según figura en el texto, el Reglamento del Parlamento Europeo y del Consejo sobre el Espacio Europeo de Datos Sanitarios (REEDS) presenta como objetivos “mejorar el acceso por parte de las personas físicas a sus datos sanitarios electrónicos personales, y el control de dichos datos, en el contexto de la asistencia sanitaria (uso primario de los datos sanitarios electrónicos), así como para otros fines que beneficiarían a la sociedad, como la investigación, la innovación, la formulación de políticas, la seguridad de los pacientes, la medicina personalizada, las estadísticas oficiales o las actividades reglamentarias (uso secundario de los datos sanitarios electrónicos). Además, su finalidad es mejorar el funcionamiento del mercado interior mediante el establecimiento de un marco jurídico uniforme, en particular en lo que respecta al desarrollo, la comercialización y el uso de los sistemas de historiales médicos electrónicos («sistemas HME») de conformidad con los valores de la Unión”. El pasado 18 de marzo se publicó una nota de la Secretaría General del Consejo de la UE dirigida al Comité de Representantes Permanentes con contenido interesante pero que no cambia nada en lo substancial. [1]
Recientemente, la Asociación por un Acceso Justo al Medicamento (AAJM) se unió a la Coalición de la sociedad civil de la Unión Europea (UE) en defensa de pacientes y ciudadanos en el Espacio Europeo de Datos Sanitarios [2] (REEDS) planteando una serie de exigencias a la propuesta del Reglamento del Parlamento Europeo y del Consejo sobre el Espacio Europeo de Datos Sanitarios [3]. Pretende este editorial ahondar más en las razones por las cuales es rechazable el texto propuesto. Quedarán cuestiones sin exponer y valorar que se realizarán en otro momento. Las organizaciones firmantes acogen con satisfacción la intención de promover el intercambio de datos sanitarios para facilitar la atención médica directa en la UE. Sin embargo, tras repasar lo más actualizado al respecto, considero que lo que se presenta como un avance para la atención médica en la UE esconde una trampa, el verdadero objetivo, que no es otro que la centralización masiva de información sanitaria de todos los ciudadanos europeos. Si bien la intención del intercambio de datos sanitarios en el marco del tratamiento médico directo de los pacientes (uso primario), puede parecer un motivo loable, es solo una disculpa para lograr los verdaderos fines denominados secundarios, que como veremos comportan riesgos muy serios contra derechos fundamentales de los ciudadanos. Para lograr el intercambio de datos sanitarios y los fines primarios propuestos existen otras alternativas que permiten la disponibilidad de la información sanitaria de los pacientes en cualquier parte del territorio de la UE, sin necesidad de los riesgos inherentes a la centralización masiva de los registros sanitarios de todos los ciudadanos europeos. ¿Por qué entonces insistir en concentrar un poder tan grande en manos de unos pocos? La respuesta es simple: control. Los datos son el petróleo del siglo XXI y la centralización de datos sanitarios abre la puerta a su explotación por parte de intereses espurios como empresas farmacéuticas, por ejemplo, para segmentar y manipular a los consumidores, compañías aseguradoras por ejemplo para discriminar en las pólizas o negar cobertura o como gobiernos para controlar y vigilar a la población.
La tecnología e-health cuenta con los algoritmos de Inteligencia Artificial (IA) y la capacidad de aprendizaje de las máquinas están permitiendo, con un desarrollo y dinamismo vertiginosos, analizar las grandes bases de datos (Big Data), que a su vez se multiplican exponencialmente y a buen seguro, suponen una auténtica revolución en la mejora de la calidad de vida desde el diagnóstico precoz y acertado al descubrimiento de tratamientos más eficaces. Pero también, y con toda seguridad, pueden constituir un elemento extraordinariamente peligroso en manos de intereses ajenos al bien social y al respeto de los derechos individuales y de las minorías. Pueden analizar con idéntica eficacia y rapidez instantánea millones de datos clínicos muy sensibles que determinen la vida de las personas y sobre todo contra sus derechos.
Es evidente que los registros planteados por el REEDS constituyen una mina de datos inagotable sobre salud y enfermedad, pero incluyen también información sobre comportamientos, costumbres, expectativas de vida, o dotación e información genéticas de la población europea. Componen, en suma, una fuente de poder extraordinaria, con valor económico y político ilimitado, para quienes puedan explotarlos, aunque con ello se puedan sacrificar o poner en grave riesgo libertades y derechos individuales y colectivos de todos los ciudadanos de la UE, así como de su descendencia. Leído el borrador, y aceptarlo es tanto como entregar la intimidad de todos los europeos, en nuestro caso de los españoles, la de nuestros hijos y su descendencia (datos genéticos) a un Gran Hermano desconocido, que no solo no supondrá beneficio alguno para los verdaderos dueños de la información, sino que puede volverse en algún momento de sus vidas contra sus derechos e intereses.
Las nuevas tecnologías pueden mejorar la calidad de la atención a los pacientes, pero la calidad depende también de otros factores tan importantes como la confidencialidad y el derecho a la protección de la intimidad, primordiales e inherentes a la dignidad de cualquier ser humano, los cuales además son inherentes al buen ejercicio de las competencias de los profesionales sanitarios y son la garantía de una atención de calidad y de un correcto ejercicio médico. Conviene recordar que el secreto médico es una promesa de silencio singular integrada en la práctica médica que es un deber médico y un derecho fundamental del enfermo, al garantizar la confidencialidad del hecho asistencial. Bajo su compromiso, el paciente puede revelar la información que considere, confiando en la lealtad del médico. El secreto médico se recoge, acumula y guarda en la historia clínica. Por ello, esta es intrínsicamente confidencial, al guardar hechos sensibles de lo más íntimo e cada paciente, el cual, con el tiempo, hasta puede olvidar, pero de la que el médico, el equipo asistencial y el Sistema de Salud son depositarios y responsables.
La concentración de datos sanitarios de toda la UE que plantea el REEDS, en caso de mala utilización pérdida o robo, pone en riesgo definitivo y no restituible la intimidad de todos los ciudadanos europeos, al poder ser utilizada en contra de sus intereses en función de sus características, circunstancias vitales, ciclo evolutivo o cualquier otro acontecimiento esencial. Daña el principio de autonomía del ciudadano si se utiliza sin su consentimiento explicito, incluso sin su conocimiento. Esta desconsideración del derecho de los ciudadanos a tener guardada su intimidad ataca frontalmente el sostén de su libre albedrío. No hay razón en beneficio de los pacientes que aconseje concentrar en registros toda la información sanitaria pudiendo identificar a cada uno de ellos y sus relaciones. Y no lo es al existir otras opciones que permitirían el acceso a toda la información de cada paciente en caso de necesidad para su atención directa. La creación del REEDS, con la centralización de sistemas de información sanitaria, es fruto del pensamiento tecnocrático y utilitarista, unido a los actuales criterios de globalización con la entrada descarada del mercado y los intereses de multinacionales y fondos de inversión en los sistemas sanitarios.
Mientras ocurre todo esto, sorprende que en España no sea posible aún disponer de toda la información sanitaria de un paciente, fuera de la comunidad autónoma de residencia. Menos aún que esa posibilidad reciproca sea posible entre los servicios sanitarios públicos y privados. Esto es un hecho que interpela a la inteligencia, a la vez que arroja dudas sobre la verdadera intencionalidad de la creación del REEDS. ¿Cómo es posible que el objetivo denominado primario que se pretende lograr, con la disculpa de la libre circulación de pacientes en la UE, nuestras Administraciones sanitarias lo hayan impedido dentro de nuestras fronteras? ¿Cómo es posible esa disponibilidad mutua entre pública y privada si no es posible dentro de nuestro espacio nacional?
Confunde y sorprende la imposición de integrar las diferentes informaciones provenientes de diferentes países y fuentes. En el caso de España, donde conviven sistemas sanitarios públicos autonómicos, concertados (MUFACE; MUJEJU, ISFAS…) y privados, obligarían a todas esas informaciones a tener la misma arquitectura y estructura para que todas estén sitas en una base de datos común y que puedan ser utilizadas posteriormente en contrapartida por cada sistema de procedencia. Pero además en España cada sistema público de información autonómico funciona, y en ellos se superponen datos de dos modelos complementarios, con dos arquitecturas y estructuras funcionales diferentes, como muy bien explicaban hace tiempo Ruiz Téllez y Alonso López (4). Por un lado, el de la Atención Primaria (AP), experta en sanos, que trabaja con sistemas y procesos diagnósticos de alta sensibilidad. Y el de Atención Especializada (AE), que trabaja solo con enfermos y procesos diagnósticos de alta especificidad, fundamentalmente con enfermedades y procesos, casi siempre con mono episodio y de forma transversal solo con el individuo enfermo y a demanda de este. Es evidente que un modelo que no contemple esas diferencias es un mal modelo que impediría el análisis y adaptación de ambas formas de actuación.
Toda la información de la que dispondrá el REEDS en las condiciones que propone y que trabajara eficazmente la IA, aun siendo enorme, diversa y heterogénea, es de tal sensibilidad e incluso especificidad que es posible desagregarla para cada uno de los millones de individuos, familias o comunidades incluidas en ella. Es extraordinariamente atractivo e incalculable el valor de tales acopios de información sanitaria. Puesto que la seguridad absoluta de los sistemas de protección no existe, una vez obtenida, en caso de robo, hackeo o cualquier otra desviación, en un instante puede trasladarse a cualquier parte del mundo en un corto espacio de tiempo. En esas situaciones, posibles las características de la información sanitaria la hacen irrecuperable, ni posible invalidar o anular, pero sí replicar, toda o en parte, tantas veces como se desee. Por ello, aunque se afirme o intente demostrar lo contrario, la propuesta ofrecida rompe con el derecho de los pacientes, ciudadanos y pueblos al control del acceso a su información sobre de su intimidad, que quedara al descubierto atentando contra su dignidad como personas.
Nos preguntábamos al principio ¿por qué entonces insistir en concentrar un poder tan grande en manos de unos pocos? La respuesta es simple, quien tiene el poder desea más poder en forma de control. Los datos son el petróleo del siglo XXI, y la centralización de datos sanitarios abre la puerta a su explotación por parte de intereses espurios como empresas farmacéuticas o compañías aseguradoras, por ejemplo, para discriminar en las pólizas o negar cobertura o gobiernos para controlar y vigilar a la población.En consecuencia, la centralización de registros y objetivos secundarios del denominado REEDS no es aceptable, porque además de los riesgos mencionados, derivados de la concentración de registros sanitarios de todos los ciudadanos de la UE, es innecesaria para que cualquier paciente pueda recibir atención sanitarias directa en cualquier parte de la UE.
El día 12 de marzo el Consejo de Ministros autorizó al Centro para el Desarrollo Tecnológico y la Innovación (CDTI) para que, a través de Innvierte Economía Sostenible SICC S.M.E., realice un pacto negociado para la constitución de una sociedad de terapias avanzadas con capital público-privado. Este desarrollo se encuentra enmarcado en el Proyecto Estratégico para la Recuperación y Transformación Económica (PERTE) para la salud de vanguardia, con la aplicación de fondos del Plan de Recuperación, Transformación y Resiliencia.
La sociedad público-privada creada se dedicará a la investigación, desarrollo y comercialización de medicamentos en nuevas terapias avanzadas.
La aportación inicial de capital público-privado contará con una cantidad aproximada de 74 millones de euros. La participación privada la realizarán las compañías farmacéuticas Rovi e Insud Pharma, y alcanzará el 51 % del total. Un 49 % del capital corresponderá a la financiación pública del Ministerio de Ciencia, Innovación y Universidades, a través de CDTI y de su sociedad de inversión Innvierte Economía. El gobierno podría aumentar su aportación a la empresa en un futuro.
La información actualmente disponible proviene de las notas de prensa proporcionadas por el CDTI (1), el Ministerio de Ciencia, Innovación y Universidades (2), así como la compañía farmacéutica Rovi (3) y de breves comentarios en la prensa económica, m-Erica y farmacéutica. Por lo tanto, a día de hoy, disponemos de datos incompletos sobre las características del acuerdo firmado. En las citadas notas se recoge la afirmación de la ministra de Ciencia: “También se pretende capacitar a empresas españolas en este ámbito, en el que todavía no tenemos industria nacional para este tipo de terapias”.
Como objetivo adicional para la sociedad, la Sra. Morant añade: “Ofrecer los fármacos resultantes de este proceso al Sistema Nacional de Salud con un precio competitivo, haciendo que nuestro Sistema Nacional de Salud sea más sostenible y facilitando el acceso a tratamientos que tienen un coste muy elevado”.
Finalmente, la ministra ha añadido que esta nueva empresa tenía como estrategia, 0desarrollar y comercializar productos originados en los resultados de grupos de investigación españoles e impulsar la industria nacional en el campo de las terapias avanzadas, disponiendo también del apoyo del Instituto de Salud Carlos III.
De esta limitada información inicial solo podemos afirmar que su dedicación principal es la investigación en terapias avanzadas, la mejora en el acceso del SNS a estos medicamentos y procedimientos, y la producción y comercialización de aquellos productos y procedimientos que finalmente se desarrollen.
Esta iniciativa, en principio, es positiva. Pero, con la información disponible, quedan algunas preguntas en el aire: ¿Quién será titular de los derechos de propiedad intelectual? ¿Cómo se fijarán los precios? ¿Se facilitarán a otras empresas y países, a través de la OMS, la licencia y la transferencia del know how, como se hizo con los test para la COVID-19?
Hemos considerado útil exponer algunas propuestas y sugerencias, que pensamos podrían ser útiles para un desarrollo más avanzado de este proyecto en líneas y aspectos que creemos clave para el futuro.
Reflexiones, consideraciones y sugerencias
Las estrategias de Public-Private Partnership (PPP) o consorcios que involucran a múltiples organizaciones de partes interesadas, incluida al menos una organización sin fines de lucro (por ejemplo, gobierno, universidad o fundación) y al menos una organización con fines de lucro (por ejemplo, empresa farmacéutica, biotecnológica o de dispositivos médicos) son habituales en determinados campos de la investigación y desarrollo. Esta iniciativa, por tanto, se encuentra dentro de un contexto con múltiples experiencias en este sentido dentro de la UE y de varios países. Durante la pandemia de la COVID-19, por ejemplo, se ha impulsado su desarrollo en países de bajos y medianos ingresos, como por ejemplo Sudáfrica, para facilitar el acceso a vacunas y medicamentos.
Conocemos también cómo la producción pública de medicamentos constituye una de las soluciones posibles al grave problema de equidad y accesibilidad de la población a los medicamentos y vacunas necesarios para la recuperación y mantenimiento de la salud.
Consideramos, como señala, por ejemplo, Dean Baker (4), que es imprescindible desarrollar alternativas al modo actual de financiación de la investigación y desarrollo de medicamentos mediante el sistema de monopolio de patentes. Un sistema que, como conocemos bien, esconde un porcentaje muy alto de financiación pública previa. Tenemos una evidencia demostrada en numerosos artículos e informes: financiar la investigación a través de la concesión de monopolios (patentes y exclusividades), cobrando precios excesivos de miles de euros por medicamentos, que en realidad costarían unas decenas o cientos de euros es inaceptable y provoca un insoportable aumento del gasto farmacéutico público.
Consideramos que la industria farmacéutica es un sector estratégico que exige implementar y articular políticas públicas, que integren las áreas de investigación, ciencia y tecnología, con las necesidades y prioridades en salud de nuestros ciudadanos y ciudadanas.
Consideramos que iniciar un camino que permita averiguar las fortalezas y debilidades de un sistema alternativo al modelo actual, por un lado, y por otro, incrementar las posibilidades nacionales de investigación y producción pública de medicamentos nuevos es un paso necesario y positivo. Además, facilita la creación de instrumentos para la gestión de políticas y tecnologías vinculadas con el desarrollo estratégico autónomo e independiente de nuestro país. Como afirmaba el secretario de Estado de Sanidad, Javier Padilla (5): “la presencia de lo público en la industria farmacéutica ha de servir para generar ecosistemas que lideren y dirijan la inversión biomédica de titularidad privada hacia ámbitos de investigación relevantes, generando un ente público con capacidad de acumular know how, en los procesos de investigación y desarrollo biomédico”.
Consideramos que el acuerdo debe incluir y garantizar que los derechos de propiedad intelectual correspondientes a los productos desarrollados se encuentren en el ámbito del dominio público. Al mismo tiempo, los resultados de las investigaciones y desarrollos deben estar disponibles de forma abierta y no deben estar sometidos a normas de confidencialidad.
Consideramos que el precio final de los productos farmacéuticos desarrollados, una vez comercializados, debe corresponder con los costes, debidamente auditados de la investigación, desarrollo y producción de estos con un beneficio razonable.
La experiencia del Consejo Superior de Investigaciones Científicas (CSIC), en primer lugar, poniendo a disposición de los países en vías de desarrollo los test serológicos para COVID-19 y posteriormente facilitando su prototipo de vacuna a través de un acuerdo con la OMS y Medicines Patent Pool, constituyen excelentes ejemplos a seguir para facilitar al acceso igualitario a tecnologías sanitarias para Covid-19.de terapias, vacunas y sistemas de diagnóstico.
Consideramos que en este acuerdo debe incluirse y garantizarse el objetivo de compartir la propiedad intelectual, conocimiento y datos, mediante licencias transparentes, no-exclusivas y transferencia de tecnología y know how, con una decisión clara de facilitar y conseguir un acceso universal y equitativo.
Finalmente reiterar que el compromiso con la financiación pública de la investigación de fármacos nos parece clave para asegurar y conseguir un acceso justo a los medicamentos y al mismo tiempo impedir el elevado gasto farmacéutico.
Referencias
1.El Gobierno aprueba la creación de la primera sociedad mercantil de terapias avanzadas con capital público-privado de España con la participación del CDTI a través de Innvierte SICC
2. El Gobierno aprueba la creación de la primera sociedad mercantil de terapias avanzadas con capital público-privado de España 12 de marzo 2024 Ministerio de Ciencia, Innovación y Universidades
La industria farmacéutica ha suscitado de nuevo el interés de la producción audiovisual en las grandes plataformas. Tras “Dopesick”, la serie “Medicina letal” retoma el caso de Purdue Pharma y la pandemia de adicciones a causa del consumo de su fármaco estrella, el OxyContin.
Cartel de la serie “Medicina letal” (“Painkiller”) (Imagen: Netflix)
Introducción
Aunque actualmente es el fentanilo el producto opioide que acapara el interés general como desencadenante de una verdadera epidemia de adicción en los Estados Unidos, es conocido por todos lo ocurrido no hace muchos años, también en lo Estados Unidos, en la década de los 90 y primera década de los 2000, la conocida como crisis de los opiáceos provocada por el consumo masivo del OxyContin.
En la crisis actual del fentanilo el problema se centra en el mercado ilegal norteamericano de drogas. En los años 90, no fue un producto ilegal manejado por mafias criminales. La epidemia fue provocada por el consumo de un producto legal, aprobado por el regulador (Food and Drug Administration – FDA). producido por un laboratorio respetable, recetado por los médicos y de venta en farmacias.
En principio, hay una notable diferencia entre las dos crisis, una se basa en un producto ilegal y el otro en un producto completamente legal; en el fondo, el comportamiento de unos, las mafias criminales de drogas ilegales, y otros, el laboratorio productor y distribuidor del medicamento legal, son lamentablemente muy similares. Cuando se junta la codicia y el rey de los opiáceos, en ambos casos, se desencadena una crisis de salud pública y cientos de miles de muertos.
Centrándonos en el OxyContin se pueden relatar los principales pasos que realizó el laboratorio propietario del fármaco, Purdue Pharma, que llevó a desencadenar la crisis de salud pública: no hubo I+D, sólo modificaron mínimamente un medicamento ya existente, manipularon descaradamente los resultados de los ensayos clínicos, patentaron este “antiguo medicamento” como un “nuevo medicamento”, consiguieron la connivencia del órgano regulador -FDA- para obtener su visto bueno al medicamento y a sus principales características (sólo produce adicción en menos del 1% de pacientes y su acción dura al menos 12 horas) y realizaron una campaña publicitaria agresiva envuelta en amigables peluches y repleta de mentiras dirigida a los médicos.
La serie de Netflix sobre la que trata este artículo (ojo, contiene spoiler) es una obra de ficción, pero está basada en hechos reales.
Notas sobre la serie “Medicina letal” (“Painkiller”)
“Painkiller”, la serie de Netflix presentada en España en 2023 como “Medicina letal” (1), acerca a los espectadores la historia de la empresa Purdue Pharma, responsable de la fabricación del opiáceo Oxycontin y de la posterior oleada de adicciones en los Estados Unidos. Producida por Eric Newman (serie “Narcos”) (2) y creada por Noah Harpster y Micah Fitzerman, se basa en el artículo de Patrick Radden (3) publicado en New Yorker, en el cual se destapaban “los trapos sucios” (4) de esta empresa y de sus propietarios, la adinerada familia de los Sackler. La miniserie está estructurada en seis capítulos, que comienzan siempre con las declaraciones frente a cámara de algún familiar que ha perdido a un ser querido -normalmente un hijo o una hija en plena juventud- a causa del consumo de OxyContin y, como se reitera al comienzo de cada uno de ellos, “está basada en hechos reales aunque algunos personajes, nombres, hechos, lugares y diálogos se han modificado con fines dramáticos”.
Richard Sackler (Matthew Broderick) en el laboratorio de Purdue Pharma (Imagen. Netflix)
La narración se hilvana a partir de las revelaciones que la abogada de la oficina del fiscal, Edie Flowers (interpretada por Uzo Aduba), tras descubrir el inusitado incremento de recetas de Oxycontin firmadas por los médicos, va realizando acerca del fraude de Purdue: la construcción de un negocio millonario a costa de la salud de miles de estadounidenses. Esta serie complementa la versión que sobre este fraude ya ofreció en 2021 la serie “Dopesick, historia de una adicción” (5), distribuida en la plataforma de Disney+.
Conforme va avanzando la narración de Edie Flowers, la trama gira en torno a diferentes personajes reales y otros de ficción: la propia Edie, que además ha vivido en su entorno familiar el problema de la drogadicción; el dueño de un taller mecánico de coches, Glen Kryger (Taylor Kitsch) (8), quien tras un accidente laboral es medicado con Oxycontin y se convierte en adicto al fármaco; una comercial joven, inteligente y con cierto atractivo físico, encargada de convencer a los médicos de la ventaja de recetar el fármaco a sus pacientes, Shannon Schaeffer (West Duchovny), y su jefa directa, Britt Hufford (Dina Shihabi), que la entrena en las “seductoras artes” del comercio farmacéutico; y Richard Sackler (Matthew Broderick), médico multimillonario y cabeza visible de la familia propietaria de Purdue Pharma (6,7). Precisamente, uno de los aciertos del “casting” ha sido la elección de un actor como Matthew Broderick (2) para interpretar al “villano” de la serie, Richard Sackler, haciendo que éste no resulte repulsivo a los espectadores, a pesar de su falta de escrúpulos y afán lucrativo. Este gestiona su personaje con una mezcla de arrogancia, soledad, serenidad e indefensión, a la vez que férrea autoridad frente a su familia y a los acontecimientos.
Junto a estos personajes principales figuran otros como el tío de Richard, Arthur Sackler (Clark Gregg) (9), quien puso en pie el emporio farmacéutico; directivos de Purdue como Howard Udell (Brian Markinson), encargado de ejecutar las malas artes de los Sackler para hacer triunfar la empresa; el médico Gregory Fitzgibbons (John Ales), enemigo de recetar OxyContin de forma tan alegre como hacían otros de sus colegas; el empleado de la FDA que autorizó el uso del fármaco, Curtis Wright (Noah Harpster); y el jefe de Edie y fiscal, John Brownlee (Tyler Ritter), encargado del caso, que tendrá que negociar un nefasto acuerdo judicial presionado por el Departamento de Justicia.
La historia es bien sencilla y conocida por muchos: los dueños de Purdue quisieron triunfar en la industria farmacéutica a la par que obtener pingües beneficios (10), para lo cual diseñaron un nuevo opiáceo para el dolor, el Oxycontin (hidrocloruro de oxicodona), que provocaba en la mayoría de los casos adicción a los pacientes; sin embargo, este “defecto” fue ocultado a las autoridades creando así un problema grave de salud pública. Pero cuando la denuncia de la fiscalía llevó a los Sackler a juicio, una “llamada a medianoche” desencadenó la protección de las altas instancias del Estado (Congreso, Casa Blanca y Departamento de Justicia) y el caso se resolvió con una indecente mínima condena por “error en el etiquetado de los fármacos”, aunque posteriores reclamaciones llevaron finalmente a la empresa a la bancarrota.
La serie va relatando, a través de Edie Flowers, este proceso de engaño a partir de la creación del fármaco, su distribución a través de una red insistente de comerciales, el ocultamiento de la fuerte adicción por parte de quienes consumían el fármaco, el dudoso papel de la FDA en la autorización de su comercialización, el descubrimiento judicial de la trama y el apoyo de las altas instancias del poder en Washington para evitar que el peso de la justicia cayera sobre Purdue Pharma.
Cómo se monta un negocio lucrativo
Los orígenes de Purdue Pharma se remontan al psiquiatra Arthur Sackler, quien comprendió que el futuro negocio de la salud mental no se encontraba en prácticas únicas e irreversibles como la lobotomía, sino en el tratamiento farmacológico, que podía proporcionar clientes para toda la vida. Así, por un lado, ideó un nuevo fármaco, Thorazine, que definió como “lobotomía en bote”, y por otro, adivinó el prometedor futuro del marketing, por lo que compró una industria farmacéutica y una agencia de publicidad y además se aseguró el contrato del Valium, la piedra angular de su negocio millonario. Ello le permitió amasar una gran fortuna y convertir el apellido Sackler en una marca de prestigio a través de sus jugosas donaciones a museos, escuelas y hospitales, con el afán de hacer perpetuar su nombre.
Sin embargo, Arthur Sackler falleció prematuramente a causa de un infarto. Las riendas del negocio familiar las heredó entonces su sobrino Richard (a lo largo de la serie, en una licencia fílmica y narrativa, se suceden los diálogos imaginarios entre Richard y el ya fallecido Arthur, quien se configura como la conciencia de su sobrino y le aconseja y recrimina en función de sus decisiones más o menos controvertidas o acertadas). Como comenta Edie Flowers: “Arthur creó la industria farmacéutica moderna e hizo a su familia millonaria, pero fue Richard, su sobrino, el que abrió la caja de Pandora y les hizo multimillonarios. Tardaría diez años, pero ese poder que Arthur le legó a Richard fue el principio del OxyContin”.
Arthur había dejado múltiples deudas tras fallecer, pero también el entramado de un conglomerado de empresas. El abogado de la familia les propuso que se deshicieran de todas ellas, pero Richard consideró que podría ser una gran negocio quedarse únicamente con Purdue, la empresa que fabricaba el MS Contin. Este era un fármaco a base de morfina, destinado a enfermos de cáncer en proceso terminal, a partir del cual pensó que podrían refundar el imperio familiar. Su argumento era muy sencillo: “Todo el comportamiento humano se basa en dos cosas básicas: huir del dolor y correr hacia el placer. Es un ciclo. Huir del dolor y correr hacia el placer. […] Si nos convertimos en la puerta de entrada de todo aquel que quiera huir del dolor, habremos cambiado el mundo. Terminaremos lo que Arthur empezó”.
Shannon Schaeffer (West Duchovny), una de las comerciales del OxyContin (Imagen: Netflix)
Richard Sackler creó así, en Purdue Pharma, uno de los analgésicos más potentes del mercado, pero ocultó deliberadamente que el medicamento no era seguro. La operación era sencilla: tan sólo había que modificar el fármaco ya existente, el MS Contin, consistente en un recubrimiento (contin) que se disolvía lentamente en el tracto digestivo y facilitaba la liberación del sulfato de morfina. Como la morfina solía asociarse a la muerte, Sackler propuso a los técnicos del laboratorio sustituir esta por otro opiáceo, la oxicodona, que nadie asociaría con la morfina, y por añadidura, con la muerte. El plan para sus dueños era perfecto, había nacido el OxyContin, un fármaco que ningún paciente con dolor podía rechazar; pero existía un punto débil que se ocultó, el fármaco creaba adicción, lo cual fue el origen de toda una pandemia.
Cómo una red de comerciales puede hacer triunfar un fármaco
Una vez inventado, el siguiente paso era comercializarlo, y ahí Richard Sackler acudió a las tácticas exitosas de su tío Arthur, quien estaba convencido de que el triunfo de un fármaco residía en el mensaje que se transmitía. Richard sabía cuál debía ser el mensaje del OxyContin -aliviar el dolor a todo paciente- y para transmitirlo se encargó de formar un amplio equipo de comerciales a partir de la premisa de que el vendedor era el rey: “Hay dos clases de personas importantes: los creadores y los vendedores. Son igual de importantes. Puede que los vendedores más. No existe mayor talento que aquel que estimula físicamente a una persona para sacar dinero de su cartera y entregártelo a ti”.
Glen Kryger (Taylor Kitsch), paciente adicto al OxyContin por un accidente laboral (Imagen: Netflix)
Así, reclutaron a un buen número de “graduadas universitarias, atractivas y con conocimientos médicos y las formaron para vender hasta a su madre”. Este ejército de comerciales fue enseñado, en diferentes reuniones y convenciones, en las que eran “motivadas y mimadas” (la mayoría eran mujeres) con grandes sueldos, cheques y coches de lujo. El mensaje que debían trasmitir era claro y meridiano: los médicos ignoran el dolor de sus pacientes, pero nadie tiene por qué soportar ese dolor, y para eso está el nuevo fármaco. Los responsables de Purdue aseguraban que el medicamento era eficaz, duradero y seguro, pudiéndose recetar además a un amplio espectro de la población. Y los comerciales repetían este mantra hasta la saciedad, impulsados por los pingües beneficios que obtenían por convencer a los médicos para que lo prescribieran sin descanso (a mayor número de recetas y de mayores dosis, mayores beneficios para los comerciales). Otra práctica de gran éxito fue transmitir la imagen bondadosa del fármaco, para lo cual crearon un enorme peluche azul con forma de pastilla de OxyContin, que entregaban en todas las farmacias donde se comercializaba el fármaco.
El medicamento se popularizó así rápidamente. El peligro ya estaba en la calle. Las adicciones crecieron con rapidez, de tal forma que muchos pacientes ya no sólo consumían las pastillas sino que las esnifaban directamente, creándose una corte de camellos y yonquis. Cuando esta situación fue denunciada en un informe interno por uno de los personajes principales, Shannon, la solución de la farmacéutica fue bonificarla económicamente y ascenderla para acallar sus críticas, convirtiéndola en reclutadora de nuevas comerciales.
El papel de los pacientes, adictos a su pesar
Los pacientes, en un primer momento, estaban convencidos de los beneficios y utilidad del fármaco para paliar su dolor. La serie los ejemplifica en Glen, el dueño de un taller que consume OxyContin tras un accidente laboral: “El OxyContin es increíble. Ha conseguido que vuelva a sentirme normal después de mi accidente. Desde que tomo OxyContin no he faltado al trabajo un solo día. Ahora puedo decir que disfruto cada día de mi vida. El OxyContin me ha devuelto mi vida”.
Si el fármaco no creara adicción no hubiera supuesto ningún peligro, pero los pacientes tratados necesitaban, al cabo del tiempo, solicitar dosis cada vez más altas a sus médicos. Poco a poco las colas ante las consultas y las farmacias se hicieron interminables, las urgencias de los hospitales se colapsaron con el aumento de los adictos, los delincuentes robaban pastillas a los pacientes para luego revenderlas y apareció un mercado negro del que se adueñaron los camellos, e incluso se difundió su consumo entre los jóvenes, como una moda, quienes ahora esnifaban las pastillas cual si fueran una nueva droga.
El fiscal John Brownlee (Tyler Ritter) y la abogada Edie Flowers (Uzo Aduba) buscando pruebas contra Purdue (Imagen: Netflix)
Aunque la pandemia era imparable, Purdue seguía afirmando que el fármaco no creaba adicción más que al 1% de sus consumidores. El medicamento debía tomarse cada 12 horas, pero sus efectos no duraban todo ese tiempo, de tal forma que los pacientes sufrían de síndrome de abstinencia dos veces al día. El OxyContin, como señala Flowers, se había revelado como un fármaco muy deficiente en los diferentes ensayos clínicos, pero eso también se ocultó: “A Richard le daban igual las deficiencias. Sólo le importaba demostrar que era eficaz suprimiendo el dolor. Y comenzó con pequeños ratones. Los siguientes ensayos fueron con humanos. En esa fase tienes que demostrar la eficacia del medicamento. Si no es eficaz, no se aprueba. Los primeros ensayos con humanos se realizaron en Puerto Rico con mujeres sometidas a cirugías ginecológicas. En la segunda fase se hicieron pruebas en Estados Unidos con pacientes de cáncer. Y en la tercera fase, Purdue llevó a cabo ensayos a gran escala por todo el país con más de mil pacientes voluntarios. Cuando revisé los ensayos, dos cosas llamaron mi atención. Una, fueron un completo desastre. Al dejar los opioides, los ratones sufrían síndrome de abstinencia, igual que los humanos. Bruxismo, dolor abdominal y temblores. En los ensayos con humanos, el OxyContin debía durar 12 horas. En Puerto Rico, la mitad de las mujeres necesitó más medicación para pasar la noche. Un tercio de los 164 pacientes de cáncer abandonaron el ensayo. No resultó eficaz, sufrieron efectos adversos o infringieron el protocolo. No informaron de ciertos incidentes. Se produjeron abandonos de pacientes en todos los ensayos. Los efectos no duraban tanto como prometían y pedían más oxicodona para pasar la noche. Era un fármaco eficaz, como les digo, pero con muchas deficiencias”.
La situación se había descontrolado, pero había porque poco margen de maniobra para las autoridades, ya que los médicos estaban recetando un fármaco legal, y sin delito no se podía actuar en contra de la farmacéutica.
Cómo algunos médicos pueden contribuir al desastre
Los médicos, que tradicionalmente recetaban fármacos como Vicodin, se rindieron al nuevo fármaco ante las campañas de marketing de Purdue. Algunos de ellos lo recetaban convencidos de su utilidad, pero otros lo hacían seducidos por la juventud y el atractivo de las comerciales. La fórmula era sencilla, como recuerda Shannon a una nueva comercial, refiriéndose a los médicos: “Sólo tienes que acordarte de los cumpleaños y llevar dónuts y café”.
Aun así, algunos médicos -pocos al parecer- rechazaban prescribirlo a discreción y sólo lo hacían cuando era estrictamente necesario, convencidos de su peligro, aunque ello les supusiera un enfrentamiento con las comerciales, como era el caso del doctor Fitzgibbons: “La molécula del OxyContin es casi idéntica a la de la heroína. ¿La heroína no es adictiva? Oxicodona, ese es el compuesto del OxyContin. Morfina, codeína, hidrocodona, hidromorfona, diacetilmorfina. Todas son heroína. Todas vienen de la amapola del opio. Distintos nombres, la misma mierda. No tienes ni idea. Una adolescente intentando venderme un narcótico de categoría II. Eres un peligro y eres tonta. Eso te hace aún más peligrosa. Voy a pedirte que salgas de mi consulta. Eres una narcotraficante con coleta”.
La connivencia de muchos médicos con las comerciales, consciente e inconsciente, extendió su uso por el país y propagó la pandemia de adicciones. Los responsables de Purdue seguían defendiendo que OxyContin sólo causaba adicción al 1%, argumentando en su defensa la publicación de un artículo en el New England Journal of Medicine, que no era tal sino una carta al editor y en la cual su autor, un médico de la Universidad de Boston, ni siquiera defendía esta afirmación, sino que analizaba el uso de los opioides a corto plazo y en el entorno hospitalario, y no a largo plazo y sin supervisión, como estaba ocurriendo con el OxyContin. Por lo tanto, un engaño más que los propietarios camuflaron además mediante la certeza científica.
El fraude comenzó poco a poco a destaparse cuando algunos inspectores, como Edie Flowers, descubrieron la sobreprescripción del fármaco: “Lo que hizo Purdue, lo que hizo Richard Sackler, abusando de la confianza entre médico y paciente, vendiendo esperanza a gente vulnerable, es de una maldad enfermiza. Richard Sackler combinó dos de las sustancias más adictivas: la codicia y el opio”. Cuando la empresa comenzó a vislumbrar que el negocio podía correr peligro, acudió a la profesión médica para que esta les sirviera de salvaguarda. Serán ellos, los médicos, los encargados ahora de hacer la publicidad del fármaco revelando sus grandes beneficios y acusando a los pacientes (“Como médico me indigna que la gente abuse de este fármaco”); sin embargo, algunos pocos, como el ya mencionado Fitzgibbons, no entraron en este juego, sino que incluso enviaron a la farmacéutica cientos de documentos en los que se demostraban las deficiencias del OxyContin. Estas quejas y reclamaciones también fueron ocultadas deliberadamente.
Tampoco la FDA sale bien parada
Los ensayos con OxyContin en sus tres fases fueron un completo desastre, pero como en esos procesos de la investigación farmacológica no falleció ningún animal de laboratorio, los parámetros fueron considerados aceptables, a examen de los expertos de la FDA, de tal forma que el fármaco se calificó como seguro y eficaz (además, la mitad de los pacientes afirmó que les aliviaba suficientemente el dolor). Según Edie Flowers, la FDA tuvo parte de culpa en la salida al mercado del Oxycontin: “En Purdue estaban seguros de que la solicitud pasaría el trámite de la FDA sin problemas. La gente cree que esas solicitudes son revisadas minuciosamente por un equipo de expertos pero ni de broma es así. Casi nadie sabe que la FDA no es más que una pequeña agencia gubernamental que suele fiarse de la palabra de las empresas. Ellos nunca realizan pruebas a los nuevos productos. Revisan lo que se les envía. En el caso de los fármacos, es una sola persona la que suele hacerlo. La persona que revisó la solicitud del OxyContin se llamaba Curtis Wright.”
Curtis Wright (interpretado por Noah Harpster, uno de los creadores de la serie) se tomó con profesionalidad su trabajo evaluador y fue un hueso duro de roer para la farmacéutica, ya que en un primer momento lo desaconsejó por su alta peligrosidad en su calidad de opiáceo, lo cual suponía un serio obstáculo para su aprobación. Los Sackler adoptaron entonces una táctica propia de la CIA (MICE: moneda, ideología, coerción y ego), como se explica en la serie, y fueron persuadiendo a Wright de tal manera que éste acabó por aprobar la comercialización del fármaco. Curiosamente, Wright, un año después de esta aprobación, abandonó la FDA y posteriormente fue contratado por Purdue (puertas giratorias, se llama).
La aprobación de la FDA dejaba el camino expedito para el negocio de Purdue y facilitaba la explosión de la pandemia, y todo por una frase que no ha vuelto a repetirse nunca más en la aprobación de ningún otro fármaco: “Se cree que la absorción retardada de los comprimidos de OxyContin reduce la probabilidad de abuso del fármaco”. La aprobación se basaba fundamentalmente no en una afirmación categórica sino en una suposición, la que respaldaban dos palabras tan poco científicas como “se cree”.
Los fiscales entran en acción
Al ser un fármaco legal era complicado actuar contra Purdue y sus directivos. El primero en hacerlo fue el fiscal de Maine, Jay McCloskey, quien envió una carta al Congreso y a más de 5.000 médicos advirtiendo del peligro del medicamento. Ante este acoso, Purdue reaccionó defendiendo su legalidad y acusó, en cambio, a los adictos, a quienes achacaba un consumo desenfrenado: “el problema son los yonquis, no nosotros”, venía a decir la empresa farmacéutica. Así, ante la muerte de una joven por consumo de OxyContin, Purdue emitió un comunicado a todas luces cínico: “La alarma social sembrada por Carol Brewster y los medios carece de fundamentos. Tenemos entendido que se encontraron múltiples sustancias en el cuerpo de la señorita Brewster, por lo que no queda claro si su muerte se debe al OxyContin o al largo y continuado historial de drogadicción de la señorita Brewster.”
Ante la carta al Congreso del fiscal de Maine, Sackler decidió tomar la iniciativa y envió a declarar a los responsables de los departamentos médico, legal y de marketing, evitando exponerse él mismo a las críticas. Los directivos se defendieron afirmando que era imposible atajar el abuso que acompañaba la comercialización del fármaco sin restringir su disponibilidad tanto a médicos como a pacientes, que necesitaban tratar su dolor; y de nuevo derivaron el problema a los adictos al fármaco al afirmar que éstos lo eran también a otras sustancias, diluyendo así su responsabilidad como empresa. Esto es, por activa y por pasiva, reafirmaban que el fármaco era seguro y que el problema era de los pacientes, que abusaban de él sin control. En su declaración ante el Congreso, negaron conocer los papeles que el doctor Fitzgibbons les había enviado denunciado los problemas del fármaco. Esta negación permitió, al final, abrir una brecha en la defensa de Purdue: habían mentido al Congreso. Parecía una burla: podrían ser condenados por mentirosos, pero no por corruptos y fraudulentos.
El fiscal jefe de Edie, John Brownlee, después de investigar los miles de papeles que Purdue les había enviado, decidió llevarles a juicio, pero antes les propuso un acuerdo amistoso: “Queremos que cambien el etiquetado para que OxyContin sólo pueda comercializarse como medicación para cuidados paliativos. Queremos que Purdue invierta en las comunidades de Virginia afectadas por su fármaco. Calculamos unos 2000 millones. Queremos una disculpa pública de Purdue. La empresa debe aceptar responsabilidades por su mal proceder.” Purdue tan sólo aceptó entregar diez millones y que en ningún momento se admitiera un mal proceder por parte de la farmacéutica.
¿En qué basaba su defensa Purdue para negarse a aceptar el pacto ofrecido por el fiscal? Consideraban que habían sido demonizados sin razón, porque OxyContin tan sólo representaba el 4% del total de opioides recetados en Estados Unidos, y porque, de todos los casos denunciados por la fiscalía, tan sólo en dos de ellos el fármaco era la única sustancia presente. Ya que los Sackler ni querían pagar ni iban a pedir perdón, Brownlee inició una nueva táctica buscando entre los afectados alguien que pudiera declarar en contra de la farmacéutica. Encontraron a Deborah, la secretaría de los Sackler, quien tenía información privilegiada al conocer los tejemanejes de la empresa y las deficiencias del fármaco, y además había sido encargada de redactar un memorándum en el que, en resumidas cuentas, se recogía que el fármaco era nefasto y muy adictivo. Y ella estaba dispuesta a declarar, sin embargo, se presentó un problema irresoluble, Deborah era adicta al OxyContin y su testimonio carecía por tanto de validez.
Pero de nuevo la fiscalía vislumbró la luz cuando Shannon, una de las comerciales, arrepentida de su forma de trabajar, decidió declarar en contra de Purdue: “Yo creía que ayudaba a la gente. Al principio era así. Créame, lo pensaba de verdad. Y después me di cuenta de que no. […] El puto dinero te hace perder la cabeza. Perdí la cabeza. Ojalá pudiera dar marcha atrás y hacer las cosas de otra manera. […] Pero ya estoy harta. No pienso hacer daño a más gente. Se acabó.” Shannon aportó miles de correos y explicó a la fiscalía cómo actuaban y cómo convencían a los médicos para que recetaran el fármaco. Con toda esta nueva información el fiscal prosiguió su lucha directa contra Purdue. Su fórmula era denunciar no a los Sackler, sino a los “lugartenientes”, esto es, a los responsables de los departamentos médico, legal y de marketing. Las razones de la acusación: mentir sobre las propiedades de un fármaco, comercializar un fármaco con mentiras, conspirar para ganar dinero y mentir conscientemente ante el Congreso al afirmar que desconocían los problemas adictivos del OxyContin.
Una llamada a medianoche
Ante el acoso de la fiscalía, Richard Sackler decidió utilizar su dinero para contratar a los mejores abogados del país: Mary Jo White, la primera fiscal mujer del distrito sur de Nueva York; Howard Shapiro, director jurídico del FBI; y Rudolph William Louis Giuliani, quien fuera alcalde de Nueva York entre 1994 y 2001. Edie estaba entusiasmada con el cariz que iban tomando los acontecimientos y con la posibilidad de acabar con Purdue de una vez por todas: “Las incontables horas y los momentos de desesperanza estaban a punto de dar sus frutos. Purdue había mentido ante el Congreso. Íbamos a por Friedman, Udell y Goldenheim [los tres responsables de los departamentos]. Se enfrentaban a muchos años de cárcel. No íbamos a pactar. Los bufones serían los primeros en caer. Y el siguiente sería el doctor Richard Sackler. Si mientes, haces daño a la gente y te dejas llevar por la codicia, al final caes. Y tienes lo que te mereces. Justicia.”
Sin embargo, para su sorpresa el fiscal Brownlee fue presionado por el más alto poder y se vio obligado a aceptar un acuerdo nefasto para la sociedad, para los pacientes y para la credibilidad de la justicia: Purdue se declaraba culpable del etiquetado fraudulento. Nada más. La razón de tal acuerdo la ofrece la serie a partir de una serie de llamadas en las altas instancias del poder. Los Sackler llamaron a Rudolph Giuliani (11), éste llamó al Congreso, desde el Congreso llamaron a la Casa Blanca, de la Casa Blanca al departamento de Justicia, y desde Justicia al fiscal Brownlee. En consecuencia, como resume Edie: “A Brownlee no le quedó otra que aceptar el acuerdo. Y así es como acabó el puto juego. Aunque mueran medio millón de personas, tipos como Richard Sackler hacen una llamada en mitad de la noche y todo sigue igual. Lo que no cambia es la cuenta bancaria de Purdue, que sigue engordando más y más. […] Lo solucionaron sin despeinarse. Purdue ganaba 30 millones de dólares a la semana. Y lo peor de todo, lo peor de todo es que nadie les paró los pies. Podían seguir fabricando Oxycontin” […] no siempre ganan los buenos”.
En resumidas cuentas, y tras el juicio donde sólo fueron acusados de etiquetado incorrecto, se sucedieron diferentes demandas contra Purdue Pharma, que acabaron por llevarles a la quiebra, y los Sackler tuvieron que admitir mala praxis y acceder a pagar 4.500 millones de dólares (12) a lo largo de diez años (dinero que obtendrían fácilmente, como señala Richard Sackler, con el rendimiento de sus inversiones y sus intereses, sin que ello les supusiera por tanto un gran quebranto), con el añadido ventajoso de que jamás nadie en el futuro les podría demandar por el OxyContin.
Los seis capítulos de la serie terminan con un texto impreso un tanto descorazonador: “Se calcula que más de 300.000 personas han muerto en Estados Unidos en las dos últimas décadas por sobredosis de opiáceos recetados como OxyContin. Cada día mueren 40 personas en los Estados Unidos por sobredosis de opiáceos. La quiebra de Purdue sigue pendiente después de revocar el primer acuerdo. No hay ningún proceso penal contra ningún Sackler por muertes por Oxycontin. Se cree que la fortuna de los Sackler supera los 11.000 millones de dólares”. “No siempre ganan los buenos”, como sentencia Edie Flowers.
En definitiva, montar un negocio lucrativo fraudulento, aunque social y saludablemente pueda ser inaceptable y hasta mortal, puede resultar altamente beneficioso si no se tienen escrúpulos y si además se cuenta con la protección de las altas instancias de la política.
5.- José Manuel Estrada, Serapio Severiano. “Los malos de la película”: la industria farmacéutica vista por el cine. Revista Acceso Justo al Medicamento. 2023; nº 17, pág. 19-25
En la duda es en la atmósfera donde debe desarrollarse la ciencia; y en la provisionalidad de los conocimientos y en las discusiones abiertas, sin embargo es la certeza y el utilitarismo lo que se impone, porque los progresos científicos estimulan la evolución social y, en el caso de la medicina, las innovaciones médicas se aceptan con facilidad porque vienen respaldadas por el prestigio profesional, porque la innovación se presenta como superior a lo reemplazado y porque la innovación, cualquier innovación, es consecuente con los valores culturales predominantes, especialmente con la poderosa ideología del culto a la salud en las sociedades modernas.
El súmmum de la fuerte e incontestable penetración de innovaciones se produce con las vacunas, donde a todas las novedades se le atribuyen solamente ventajas y donde las dudas razonables se rechazan; se etiqueta de negacionistas a los que dudan y se les ignora, y se representa de forma negativa, denigrante y demonizadora a las personas que vacilan del relato oficial, supuestamente científico, sobre las nuevas vacunas, o sobre todo producto que se comercialice con el apellido de vacuna.
No siempre fue así. Hubo un tiempo de consenso sobre la efectividad de las vacunaciones esenciales o sistemáticas de la infancia (poliomielitis, difteria, tétanos, tosferina, sarampión, rubeola, parotiditis), porque la razón entre beneficios respecto a efectos adversos, inconvenientes y costes era claramente superior a la unidad, por el contrario, con algunas de las vacunas recientemente introducidas no se puede mantener con tanta seguridad que esa relación sea claramente favorable al numerador.
Para conocer la efectividad de una vacuna no basta con obtener una titulación elevada de anticuerpos en sangre, porque su efecto epidemiológico depende del porcentaje de individuos realmente protegidos entre los vacunados, o tasa de protección, la cual suele ser inferior al 100 %, y del porcentaje de vacunados entre la población a vacunar, o tasa de cobertura de la vacunación, que varía en función de la intensidad del programa vacunal, y de la propia calidad de la vacuna, y del tipo de vacuna en sí (si la protección es individual, como en el caso del tétanos, por ejemplo, o si altas tasas de cobertura con una vacuna de buena calidad pueden cortar la cadena de transmisión, caso del sarampión); además de la propia distribución epidemiológica de la enfermedad.
Los métodos utilizados para medir la efectividad clínica de las vacunas son la comparación de las tasas de incidencia, los estudios seroepidemiológicos y la evaluación epidemiológica sobre el terreno, mediante los estudios de poblaciones, o las encuestas en las epidemias, o las tasas de ataque secundario en familias o en racimo, o la determinación de la cobertura vacunal y/o los estudios casos-control. Mención aparte merecen los métodos para evaluar la eficiencia: estudios coste/ beneficio y estudios coste/ eficacia.
Algunas vacunas motivo de controversia son, por ejemplo, la vacuna contra el herpesvirus papiloma (VHP). En España el cáncer invasivo de cuello de útero es una enfermedad rara, con tasas de mortalidad bajas, y se cuenta con una prueba de detección eficaz: el test de Papanicolau. La eficacia de la vacuna es controvertida, porque no previene de todos los serotipos de VHP relacionados con el cáncer, porque la infección no conduce irremediablemente al cáncer, porque no hay seguridad del tiempo de duración de la inmunidad en los serotipos que la componen, porque puede haber efectos adversos graves en niñas sanas, incluso muertes e incapacidades de por vida, cuando esas mismas niñas podrían vivir sanas y, de no vacunarse, en la edad adulta realizarse un test de Papanicolau más inocuo y eficaz. Es más, puede ser peligroso que la falsa sensación de seguridad que da la vacuna haga disminuir la cobertura de las campañas de detección con Papanicolau, por otra parte, mucho más baratas que la vacuna.
Así pues, la inclusión de la vacuna contra el VHP en el calendario vacunal se realizó sin que existieran pruebas sólidas y unánimes a su favor, en medio de pronunciamientos científicos y profesionales controvertidos. Fue una decisión administrativa sobre una actividad preventiva que no vale lo que cuesta, porque no estaba demostrado que fuera suficientemente útil ni suficientemente eficaz. Actualmente sigue habiendo muchas dudas sobre su eficacia (sigue habiendo casos en mujeres vacunadas) y seguridad (efecto neurológico del uso de adyuvantes de aluminio, por ejemplo) y sobre distintas irregularidades farmacológicas y comerciales.
En otros países, como en Japón, se retiró la vacuna a poco de su introducción en 2013 debido a las reclamaciones de los padres por los efectos adversos observados. Nueve años después se ha vuelto a reintroducir porque se observaron más casos de cáncer invasivo de cuello de útero en las cohortes de niñas no vacunadas. Además, se intensificó la realización del test de Papanicolau en esas cohortes. Es muy difícil retirar una medida que se ha propuesto como necesaria, pero se puede hacer, reevaluar y recuperar (si se considera que la medida es eficaz), como demuestra el caso de Japón.
En 2015, cuando se decidió la introducción en el calendario vacunal de la vacuna de la varicela, el grupo GRADE-Evalmed estudió la mortalidad y las hospitalizaciones por varicela y por herpes zóster en Navarra, Madrid, Ceuta y Melilla, donde ya se administraba la vacuna, comparando las tasas con el resto de España. No encontraron diferencias estadísticamente significativas en ninguno de los tramos de edad analizados entre el período anterior (1999–2004) y el período posterior a la vacuna (2005–2013) en ninguna de las cuatro variables estudiadas (hospitalizaciones y defunciones por varicela y herpes zóster), tanto en los dos grupos de estrategias vacunales como en la totalidad de España, con la excepción de un único ítem: hospitalización por varicela en el tramo 0 a 4 años en las dos CCAA y dos Ciudades autónomas que vacunan de varicela a los 15–18 meses, pues hubo una tasa anual promedio de 17,01/100.000 (0,017013%) en el período posterior (2005–2013) frente a la tasa anual promedio 42,37/100000 (0,042367%) en el período anterior (1999–2005), RR 0,4 (0,23–0,7); RAR 0,0254% (0,0092% a 0,0404%); NNT 3944 (2478 a 10847) y potencia 90,84%. Estimaron tal NNT de magnitud de efecto muy baja, pues significaba que habría que vacunar entre 2.478 y 10.847 niños para evitar la hospitalización por varicela de 1 niño [1].
En el mismo sentido, algunos técnicos cualificados, como Francisco Salmerón García, alertaron de que la introducción de la vacuna de la varicela era una decisión más compleja de lo que se quería representar administrativamente; que había que tener en cuenta el problema del herpes zóster; que no había pruebas que permitieran tener cierta seguridad de que la vacunación en la primera infancia no acabara produciendo problemas más graves, como el aumento de la varicela en adultos, la varicela en embarazadas e incluso un aumento de herpes zóster en la población mayor [2]. Aunque la incidencia de herpes zóster está aumentando, dicho incremento se achaca al envejecimiento de la población.
En su momento, la decisión de incluir la vacuna contra la meningitis B en el calendario vacunal fue objeto de reparos, porque debido a la relativa baja incidencia de la enfermedad, la eficacia cuestionable en el tiempo de esta vacuna y su elevado precio, no parecía lógico incorporarla al calendario vacunal ordinario, pero sí emplearla de manera hospitalaria para aquellas personas que, de manera individual por situaciones de inmunodeficiencia, la necesitasen. Por ejemplo, la Sociedad Española de Salud Pública y Administración Sanitaria (SESPAS) se mostró partidaria de circunscribir la vacuna de la meningitis B a los grupos de riesgo porque la inclusión de la vacuna frente a meningitis B en el calendario de alguna Comunidad Autónoma parecía basarse en criterios más propios del ámbito político que del científico [3]. El antes citado Francisco Salmerón denunció la captura de la política sanitaria y de las sociedades científicas a propósito de la presión ejercida por la industria comercializadora de las vacunas de la varicela y de la meningitis B [4].
Nirsevimab es un anticuerpo monoclonal, aunque se está comercializando desde 2023 como si fuera una vacuna contra la infección por virus sincitial respiratorio. El grupo GRADE-Evalmed ha analizado el ensayo clínico Harmonie. Encontraron que, de cada 115 bebés participantes: a) 113 no sufrirían hospitalización por enfermedad no grave durante los 80 días en ambos grupos (Nirsevimab vs grupo control); b) 1 tendrá el evento tras 33 días en ambos grupos; y c) 1 evitará el evento durante los 80 días en el grupo de Nirsevimab, mientras que ese 1 sufrirá el evento tras 40 días en el grupo de ninguna intervención. En cuanto a la hospitalización por enfermedad grave, de cada 287 bebés participantes: a) 285 no tendrán el evento durante los 80 días en ambos grupos; b) 1 tendrá el evento tras 27 días en ambos grupos; y c) 1 evitará el evento durante los 80 días en el grupo de Nirsevimab, mientras que ese 1 sufrirá el evento tras 40 días en el grupo de ninguna intervención. Los efectos adversos fueron desfavorables para el grupo de Nirsevimad. La dosis de Nirsevimad cuesta 209 € [5]. Un balance nada optimista.
Y llegamos a las vacunas contra el coronavirus SARS-Cov2 sobre las cuales se acumulan en apenas cuatro años, millones de artículos y de opiniones distintas, de manera que es difícil encontrar un camino de razón para conocer la efectividad de dichas vacunas [6].
Con los datos disponibles, parece que la inmunidad natural es superior a la adquirida por vacunas [7]. También, según Fraiman, Erviti et al. se ha detectado un exceso de riesgo de eventos adversos graves en los ensayos clínicos publicados sobre las vacunas ARN de Pfizer y de Moderna, lo cual apunta a la necesidad de análisis formales de daño-beneficio, particularmente de aquellos que están estratificados según el riesgo de resultados graves de COVID-19, lo cual necesitaría la publicación de conjuntos de datos a nivel de participante [8].
En los 28 días posteriores a la vacunación con la vacuna de Pfizer se produjo un incremento de la tasa de episodios de enfermedad coronaria aguda (RR, 1.13 [95% CI, 1.02-1.25]), enfermedad cerebrovascular (RR, 1.09 [95% CI, 1.05-1.13) y de trastornos de la coagulación (RR, 1.12 [95% CI, 1.07-1.19]). En el caso de Moderna el incremento del riesgo se produjo en enfermedad cerebrovascular (RR 1.21 [95% CI, 1.09-1.35]) y trastorno de la coagulación (RR, 1.26 [95% CI, 1.07-1.47]). La vacuna Astra Zéneca tenía riesgos incrementados en todos los tipos de trastornos analizados. También se han comunicado brotes de herpes zóster [9]. Los efectos adversos conocidos son muy variados, seguramente infraestimados por la escasa costumbre de comunicarlos, por la minusvaloración del riesgo por parte de profesionales y pacientes, o por la distancia temporal entre el efecto y el acto vacunal lo que dificulta poner en relación ambos acontecimientos.
Finalmente, hay dudas sobre la relación de la vacunación contra la COVID-19 con la sobremortalidad en los años 2021 y 2022. Hay datos que invitan a comprobar esta hipótesis. Por ejemplo, en un informe de situación del sistema MoMo, del Centro Nacional de Epidemiología perteneciente al Instituto de Salud Carlos III, que vigila el exceso de mortalidad por todas las causas [10], se podían comparar dos períodos prácticamente idénticos que engloban los meses de agosto de 2020 y 2021. El período de 2020 abarca del 20/07/2020 al 29/08/2020. En este periodo, se observó un exceso de mortalidad del 10,5 %: 7,8 % para hombres, 13,2 % para mujeres, 5,1 % para < 65 años, 5,0 % entre 65-74 años y 12,7 % para mayores de 74 años. En el período de 2021 (del 19/07/2021 al 30/08/2021) la sobremortalidad era bastante superior a la del mismo período de 2020 en todos los grupos: 15,8 % de sobremortalidad total, 16,1 % para hombres, 15,8 % para mujeres, 8,2 % para < 65 años, 15,0 % entre 65-74 años y 17,9 % para mayores de 74 años, siendo mayor la brecha entre los 2 períodos en las edades más avanzadas (destaca el grupo 65-74 años), donde prácticamente toda la población está vacunada y habiendo fallecido en 2020 una parte importante de los más débiles.
En una CCAA que comunicaba datos diarios de fallecidos por COVID-19 (Extremadura) también se observó que en 2021, con elevadas tasas de población vacunadas, hubo un exceso de hospitalizaciones y de fallecidos en agosto respecto al mismo periodo de 2020. En el caso de los fallecidos, 7 en 2020 y 69 en 2021, el 84 % vacunados [11]. En cuanto a los fallecidos diarios atribuidos a COVID-19 en toda España, comparando una vez más los datos de agosto de 2020 con los de agosto de 2021, en agosto de 2020 se notificaron 752 muertes por COVID-19, y en agosto de 2021, se contabilizaron 2.207 fallecidos [12].
La mayoría de los países desarrollados vienen observando excesos de mortalidad desde 2021, en Reino Unido, en Alemania y en todos los países incluidos en el EuroMOMO entre 2019-2023. El caso más singular es la mortalidad en el grupo de 0 a 14 años, ya que en el 2021 y 2022 las muertes por encima de las esperadas son 719 y 1168 respectivamente. La cuestión es si las vacunas y sus fechas de administración podrían ayudar a explicar esa evolución [13].
Lo deseable de una vacuna, también de las vacunas contra la COVID-19 es que: 1) Indujeran una inmunidad persistente, incluso de por vida; 2) Disminuyeran los casos, las hospitalizaciones y las muertes por COVID-19; 3) Produjeran escasos y leves efectos adversos a corto y largo plazo; 4) Cortaran la cadena de contagios al impedir que el virus SARS-CoV-2 se alojara y proliferara en las mucosas de los vacunados (con lo que protegerían a quienes no se pueden vacunar, lo que llamamos inmunidad de rebaño); 5) No provocaran la evolución del virus en el sentido de forzar la generación de mutaciones más contagiosas y letales. Para verificar todo esto se necesita una vigilancia exhaustiva de toda la cadena de elaboración, comercialización, administración y seguimiento temporal de la vacuna. Puede que a la industria fabricante le baste con elaborar y vender, pero a las administraciones sanitarias les debería preocupar más el efecto sobre el terreno de dichas vacunas, y poner los instrumentos y los técnicos y los sistemas de información necesarios para averiguarlo.
Por ejemplo, en el caso de los niños se deberían realizar estudios de seroprevalencia de anticuerpos debidos a infección en la infancia y postvacunales, porque debemos estar seguros de la bondad y seguridad de una vacuna, porque estamos vacunando a niños sanos y estamos poniendo en riesgo la confianza en la vacuna y en el conjunto de las estrategias de vacunación. Otro tanto debería exigirse para que se comparen las incidencias entre vacunados y no vacunados, para que se comparen poblaciones, y para que se evalúe su eficiencia en función de coberturas y costes.
Los poderes públicos tienen la responsabilidad de exigir mejores productos, por su poder monopsónico, por su poder de demanda centralizada y por sus responsabilidades de salud pública. Lamentablemente los gobiernos de la mayoría de los países se han entregado al impulso de la alta tecnología, no toleran la incertidumbre cuando sus sociedades empujan hacia la adopción inmediata de medidas sanitarias que les otorgan el consuelo de la supuesta certeza, porque vienen respaldadas por la industria, por algunas figuras prestigiosas, frecuentemente inundadas de conflictos de interés, y por las sociedades científicas con sus consensos y sus medios de financiación y formación frecuentemente vicarios de la industria farmacéutica.
Los sistemas sanitarios están perdiendo la partida frente a las multinacionales farmacéuticas. Los sistemas sanitarios no se han reforzado para ser capaces de evaluar con rigor las nuevas vacunas y su eficacia sobre el terreno, incluso han aceptado vacunas basadas en ensayos clínicos metodológicamente deficientes en el caso de la vacuna frente a la COVID-19, pero la industria farmacéutica ha asentado su poder adueñándose de las primicias tecnológicas e imponiendo sus condiciones en la metodología científica, condicionando el acceso a los nuevos medicamentos y multiplicando sus ganancias con las vacunas.
Berner-Rodoreda, Frank Cobelens, Anne-Mieke Vandamme, Günter Froeschl, Jolene Skordis, Elil Renganathan, Ellen t’Hoen, Mario Raviglione, Albrecht Jahn, Till Bärnighausen.
Este articulo tiene un gran interés. En él se realiza un análisis de la propuesta de financiar a la industria farmacéutica en la investigación de medicamentos útiles para evitar la resistencia microbiana, a través de los denominados vales de exclusividad transferibles. (TEDV)
¿Qué hay detrás de estos términos? ¿Qué significan y que suponen realmente?
Las grandes empresas farmacéuticas han conseguido imponer en la Unión Europea un relato consistente en considerar que la investigación en este campo solo es rentable si recibe incentivos y compensaciones adicionales. Para ello proponen que esta financiación llegue no directamente del fármaco desarrollado, si no a través de de la prolongación del periodo de exclusividad de las patentes de otros fármacos de la empresa. Es lo que denominan “vales de exclusividad”
Se trata, como es habitual, en la estrategia de la BigPharma de intentar prolongar al máximo el periodo de exclusividad de los fármacos patentados para así mantener el monopolio y los altos precios
Este planteamiento ciertamente perverso es el que los autores estudian de forma rigurosa. Su conclusión final es clara: los vales de exclusividad no son el instrumento adecuado para conseguir antimicrobianos frente a la resistencia antibiótica… “Los TDEV implican el mantenimiento de altos costos para otros medicamentos sin garantizar que se desarrollen y produzcan nuevos antimicrobianos necesarios contra los microbios multirresistentes.”
Resumen
La resistencia antimicrobiana (AMR) es un desafío global y europeo que conduce a muertes evitables y altos costos del sistema de salud.
Con el fin de estimular las innovaciones antimicrobianas, la Comisión Europea propone el uso de vales de exclusividad de datos transferibles (TDEV) como parte integral de la reforma farmacéutica de la UE de 2023: incentivar el desarrollo de antimicrobianos mediante la concesión de exclusividad de datos en cualquier medicamento de la elección del fabricante.
Los TDEV implican el mantenimiento de altos costos para otros medicamentos sin garantizar que se desarrollen y produzcan nuevos antimicrobianos necesarios contra los microbios multirresistentes.
Los responsables políticos europeos deben considerar la desvinculación de los incentivos de los precios de los medicamentos y la oferta de una combinación de mecanismos de empuje y mecanismos de extracción para aumentar el impacto y la sostenibilidad de la reforma farmacéutica de la UE propuesta en la superación de la AMR.
Introducción
El acceso a los productos farmacéuticos empeoró durante la pandemia de COVID-19, cuando los desarrolladores clave de ingredientes farmacéuticos activos y medicamentos dieron prioridad a sus mercados nacionales. Estos desafíos llevaron a un mayor compromiso de la Unión Europea (UE) para ser más autónoma en el desarrollo y el acceso a los productos farmacéuticos para su población.1
En abril de 2023, la Comisión Europea (CE) propuso una reforma de la regulación farmacéutica de la UE para mejorar el enfoque en el paciente, fortalecer la industria farmacéutica europea e incentivar la innovación farmacéutica.1 Esta reforma debe ser aprobada por el Parlamento Europeo. En su forma actual, incluye la introducción de vales de exclusividad de datos transferibles (TDEV) para abordar la crisis de la innovación antimicrobiana. En este comentario, evaluamos el uso de TDEV como un elemento importante de la estrategia farmacéutica propuesta por la CE sobre la resistencia a los antimicrobianos (AMR).
AMR: una grave preocupación para la UE y para todo el mundo
Abordar la AMR es uno de los objetivos clave de la reforma de la regulación farmacéutica de la UE. La AMR constituye una de las amenazas para la salud más importantes de la UE, lo que lleva a 35 000 muertes al año y le cuesta a los sistemas de salud de los Estados miembros 1.500 millones de euros al año.2 La UE publicó su primer plan de acción contra la creciente amenaza de la AMR en 2011; la OMS su Plan de Acción Global en 2015,3 4 con ambos organismos discutiendo intervenciones adecuadas décadas antes de estas publicaciones. La AMR es claramente no solo una gran amenaza europea, sino un grave problema global con 10 millones de personas que se estima que morirán de AMR para 2050.5 Un análisis exhaustivo de la carga de AMR en 204 países estimó que en todo el mundo 4,95 millones de muertes se asociaron con AMR en 2019,6 con la mayor carga de AMR asociada a hospitales en los países de ingresos medios.7 Por lo tanto, las soluciones sostenibles deben ser a escala global y trascender las fronteras europeas.
En su reforma de 2023, la CE propone reducir la AMR a través de las siguientes medidas: (1) un enfoque de una sola salud, destacando (2) el uso prudente de antimicrobianos (por ejemplo, las recetas racionales para los seres humanos y la reducción de las ventas de antibióticos para los animales de granja y la acuicultura en un 50 %); (3) mejorar el acceso y la asequibilidad de los antimicrobianos; (4) la cooperación y el apoyo global del nuevo Acuerdo de Pandemia de la OMS para la prevención, la preparación y la respuesta, y manteniendo la AMR como una cuestión clave para la implementación de la Estrategia de Salud Mundial de la UE y (5) la investigación y la innovación tecnológica con los TDEV como un incentivo para el desarrollo de antimicrobianos innovadores.1 2
TDEV para antimicrobianos
La CE propone ofrecer TDEV a los fabricantes que desarrollan nuevos antimicrobianos. Los vales otorgan a los fabricantes un año adicional de exclusividad de datos sobre cualquiera de sus medicamentos y la posibilidad de vender el vale a los desarrolladores de otros medicamentos.1 La propuesta prevé una evaluación después de 15 años (ibid). La exclusividad de los datos garantiza un monopolio de mercado al prohibir a los competidores registrar un producto genérico o biosimilar. El grupo de trabajo de expertos de la OMS en investigación y desarrollo concluyó ya en 2012 que la exclusividad de los datos no contribuye a las innovaciones en los medicamentos necesarios en todo el mundo.8
Si bien la CE especifica que los vales están restringidos a los «antibicones que cambian el juego que abordan la AMR y los patógenos prioritarios reconocidos por la OMS»,1 la OMS se lamenta en su informe de 2021 de que solo 6 de los 27 antibióticos que se están desarrollando para abordar los patógenos prioritarios cumplen al menos un criterio de innovación y solo dos son activos contra las bacterias multirresistentes. El ochenta por ciento de los antibióticos «nuevos» pertenecen a clases que conducen fácilmente a la resistencia cruzada.9 Por lo tanto, la UE debe asegurarse de que los antimicrobianos de nuevo desarrollo tengan un claro beneficio clínico sobre los existentes antes de conceder al fabricante un TDEV.
Para combatir eficazmente la AMR, las drogas deben estar disponibles y usarse con prudencia, no solo en unos pocos países, sino en todas partes. Los análisis de antibióticos han demostrado que la mayoría de los nuevos medicamentos se registran inicialmente en países de ingresos más altos con disponibilidad baja y retardada en países de ingresos bajos y de ingresos medios bajos.10 Incluso en países de ingresos altos, vemos una disparidad en la disponibilidad de medicamentos. De los 18 nuevos antibióticos aprobados entre 2010 y 2019, 17 se lanzaron comercialmente en los EE. UU.; en Canadá, por el contrario, solo dos.11 De los 14 nuevos antibióticos aprobados por la Agencia Europea de Medicamentos, 10 o más se lanzaron en el Reino Unido y Suecia, mientras que en otros países europeos, muchos no estuvieron disponibles (ibid). La ausencia o el retraso de los lanzamientos comerciales puede ser atribuible al problema subyacente que ha obstaculizado el desarrollo de los antimicrobianos en general: baja rentabilidad debido a los precios generalmente bajos y los cortos períodos de uso de los antimicrobianos.
El sistema de vales es un sistema de recompensas rentable para los fabricantes, que pueden ampliar el alcance de este sistema para ampliar los monopolios de sus medicamentos financieramente lucrativos. Esto, a su vez, conducirá a aumentos de costos para los sistemas de salud y retrasará la disponibilidad de compuestos genéricos o biosimilares más baratos de estos medicamentos.12-14 Esto ya estaba previsto en 2016 en una revisión de AMR.5 En resumen, los vales «empujarían el costo del desarrollo de antibióticos a un conjunto arbitrario de pagadores y pacientes (aquellos que usan los medicamentos en los que se aplica el vale)» (ibid. Los costos sociales reales de estos vales son probablemente mucho más altos que las proyecciones en las que la UE parece estar basando su modelo.12 13
A pesar de la importancia de otras medidas propuestas por la UE, es probable que el sistema de vales aumente los precios de los medicamentos y bloquee la entrada en el mercado de los genéricos. Tampoco puede producir mucho beneficio en términos de generar antimicrobianos verdaderamente novedosos, el objetivo final del incentivo del vale. Esta es también la postura de un documento no oficial (documento oficial o documento de debate para negociar posiciones dentro de la UE) dirigido por los Países Bajos y apoyados por 13 países europeos que critican el sistema de vales por «estifilizar la innovación de los competidores y retrasar la introducción de genéricos» y probablemente traer «altos costos a los sistemas nacionales».15 Más allá de los académicos y los políticos, más críticas provienen de las organizaciones no gubernamentales y otros actores de la sociedad civil (https://medicineslawandpolicy.org/2023/03/how-not-to-to-solve-a-crisis-the-european-commissions-plan-for-transferable-data-exclusivity-vouchers/ (acceso: 17.10.2023); https://www.msfaccess.org/msf-responds-european
Ofrecer incentivos desvinculados de las ventas, pero estrechamente vinculados al valor clínico del medicamento
En vista de las preocupaciones destacadas anteriormente, el enfoque debe cambiar hacia mecanismos alternativos. Sugerimos incluir incentivos de empuje e incentivos de tracción.16 En primer lugar, la financiación inicial debe «impulsar» la investigación y el desarrollo tempranos y preclínicos, como se recomienda en el informe de Preparación y Respuesta a Emergencias de Salud (HERA)17, y proporcionar recursos adecuados para la investigación básica y clínica para AMR en general.12 14 En segundo lugar, los mecanismos de «tracción», mencionados por la CE,1 se establecen con más detalle en el informe HERA17: Incluyen el Esquema Anual de Garantía de Ingresos como un complementación a las ventas del mercado. Esto puede tener diferentes niveles de apoyo financiero garantizado de la UE en función de los beneficios para la salud pública de la innovación de medicamentos (ibid.). Otros incentivos son las recompensas de entrada en el mercado, o una recompensa basada en hitos para las fases I y II del desarrollo de medicamentos.1 17
El desarrollo de medicamentos antimicrobianos se presta a desvincular los incentivos para la innovación del precio de venta del medicamento. La desvinculación podría implementarse a través de la financiación directa y los premios de hitos y posiblemente combinarse con los compromisos de compra para asegurar las perspectivas de mercado, combinando así mecanismos de empuje y mecanismos de atracción para incentivar el desarrollo de antimicrobianos innovadores. Algunos críticos también sugieren cobrar a los fabricantes una tarifa adicional de autorización de comercialización por todos los medicamentos no antimicrobianos, lo que complementaría la financiación de entrada en el mercado para los antimicrobianos.14
Conclusión
Los responsables políticos de la UE deben revisar cuidadosamente la propuesta de reforma de la regulación farmacéutica de la CE con su importante objetivo de abordar eficazmente la AMR como una emergencia sanitaria mundial. Los pros y los contras de las posibles soluciones para incentivar la innovación antimicrobiana deben, en última instancia, ser examinados sobre la base de sus beneficios para la salud pública. Los TDEV parecen ser los menos propensos a producir los resultados deseados.
Atención lector
Las referencias bibliográficas de notable interés se encuentran disponibles en la edición original.
Recogemos esta breve nota de Right to Breathe por su contenido humano y también por el camino que muestra de resistencia ciudadana frente al terrible poder económico de la industria farmacéutica.
Algo verdaderamente histórico está sucediendo en Sudáfrica en la lucha por el acceso a los medicamentos.Cheri Nel es una paciente con fibrosis quística (FQ) de 39 años de Johannesburgo, Sudáfrica. Ella está llevando a la compañía farmacéutica estadounidense Vertex Pharmaceuticals, los fabricantes del medicamento para la fibrosis que salva vidas Trikafta, a los tribunales por negarle el derecho a la atención médica.
Este caso legal busca anular el monopolio que Vertex Pharmaceuticals tiene sobre una medicina vital pero muy cara que necesita para sobrevivir. Con un precio de 326.000 dólares por paciente al año, Trikafta tiene la promesa de extender drásticamente la esperanza de vida de los pacientes con FQ.
Si Cheri gana, se le otorgará una «licencia obligatoria», que anularía el monopolio de la patente de los medicamentos, allanando el camino para que se proporcionen versiones genéricas del medicamento que cuestan una fracción del precio de la compañía estadounidense.
Vertex se ha defendido presentando una respuesta de 800 páginas ante el Tribunal Superior que señala su intención de proteger su monopolio a pesar de la amenaza que representa para las vidas de los pacientes sudafricanos con FQ.
Pero ahora, el caso se está llevando a un nuevo nivel histórico. Cheri está pidiendo a todos los pacientes con FQ elegibles para el tratamiento en Sudáfrica que se unan a ella como co-solicitantes en este caso legal histórico, lo que significa que si Cheri gana, todos los pacientes con FQ elegibles también ganarían.
Se cree que esta movilización legal masiva es la primera vez en cualquier parte del mundo en la que los pacientes han asumido el monopolio de una compañía farmacéutica de esta manera
Cheri dijo:
Esta enorme empresa estadounidense ha utilizado sus monopolios de patentes y miles de millones en el banco para intimidar a los pacientes con FQ y a los gobiernos de todo el mundo. Han retenido nuestras vidas para un rescate mientras tratan de maximizar sus ganancias, sin preocuparse por el costo humano. Mi lucha siempre ha sido para todos los sudafricanos con CF. Ahora tenemos la oportunidad de estar todos juntos y mostrar a Vertex, en el espíritu de Ubuntu, unidos, somos más fuertes de lo que nunca serán».
Más allá de Sudáfrica, se han puesto en marcha esfuerzos para revocar el monopolio de Vertex sobre Trikafta en varios países de los cuatro continentes a medida que los pacientes y sus familias se movilizan para hacer frente a la amenaza para sus vidas derivada del comportamiento de la empresa.
Vertex tiene más de 13 mil millones de dólares en el banco, sin embargo, solo dos países de ingresos medios tienen acceso a este medicamento que salva vidas, mientras que ningún país de bajos ingresos tiene acceso. ¡Los pacientes han tenido suficiente con esta especulación sanitaria injustificable!
Este caso legal es mucho más que Sudáfrica. Incluso se trata de algo más que fibrosis quística. Una victoria legal contra Vertex en Sudáfrica tendría grandes ramificaciones para la capacidad de las comunidades de CF en otros países para seguir tácticas similares para obtener el acceso. No solo esto, sino que representaría un desafío sin precedentes para el actual modelo global de grandes farmacéuticas, que está costando vidas todos los días al tiempo que genera enormes ganancias.
Tanya, la madre de un hijo pequeño con FQ, dijo:
«Me uno a Cheri como co-solicitante en este caso en nombre de mi hermoso hijo Janco, que tiene 6 años. Ha pasado por mucho en su corta vida y, al igual que cientos de otras familias de las FQ en Sudáfrica, necesitamos acceso a estas drogas que salvan vidas ya . Al actuar juntos, realmente creo que seremos escuchados. Mi hijo se merece una vida y un futuro y haré todo lo que pueda para darle precisamente eso»
Cada uno de nosotros merece el derecho a respirar. Al unir fuerzas en este caso legal histórico, los pacientes con FQ de Sudáfrica tienen la oportunidad no solo de salvar vidas, sino también de hacer historia.
Como es habitual, Joan-Ramón Laporte, miembro de la Comisión Editorial de la revista y colaborador habitual, demuestra en esta entrevista en eldiario.es su agudeza e inteligencia en las respuestas y proporciona a las y los lectores. Reflexiones y conocimiento sobre el entramado en el que se sustenta la investigación y desarrollo de nuevos fármacos.
El catedrático de Farmacología y exjefe de servicio del Hospital Vall d’Hebron relata en esta entrevista las prácticas de la industria farmacéutica y cómo centran sus esfuerzos en que la población se sobremedique más que en encontrar curas para enfermedades: “Curar no es negocio”, sostiene.
Los tratamientos farmacológicos: curar no es negocio; No podemos confiar en la investigación publicada; Sobremedicados y polimedicados; La invención y exageración de enfermedades; El mito del colesterol. Desde el índice de su Crónica de una sociedad intoxicada (Península), Joan-Ramon Laporte, catedrático de Terapéutica y Farmacología clínica en la Universidad Autónoma de Barcelona, promete guerra. Y cumple.
Con la perspectiva de haber sido jefe del servicio de farmacología del Hospital Vall d’Hebron de Barcelona, Laporte repasa en este libro cómo se ha llegado a una situación en la que, según denuncia, la población está sobremedicada a base de prescripciones excesivas, de enfermedades “inventadas o exageradas” bajo los dictados de una industria, la farmacéutica, oscura, centrada en la venta de medicamentos y que invierte más en promocionar sus productos y formar a los médicos en la prescripción de fármacos que en investigación y desarrollos de nuevas curas.
Durante la entrevista, en la que Laporte no se calla nada, expresará sus dudas sobre cuánto de lo que relata conoce la sociedad y reivindica un sector público más involucrado en el sector farmacéutico.
Uno diría que entre la población puede que exista esa idea de que las grandes farmacéuticas priorizan el negocio sobre la salud, por decirlo suavemente, pero no sé si a nivel general se conocen las prácticas que relata en el libro.
El libro entra en cuestiones con detalle, sobre todo en relación con el fraude en el desarrollo de medicamentos. Primero, el secretismo que rodea todos los estudios de farmacología experimental. Tiene una lógica comercial, sin duda, pero después el secretismo sigue en la investigación clínica y ahí estamos hablando de experimentar con seres humanos. Además, se adulteran los datos y se manipulan. En el libro intento explicar cómo los datos de la investigación clínica son convenientemente manipulados para dar una apariencia del fármaco que no es la realidad en cuando a su eficacia y seguridad. Las acciones de todos los fármacos son muchas aunque nos vendan solo una, como si fuera una bala mágica diseñada específicamente para ejercer una acción y solo una acción. Los resultados de la investigación clínica no es que tengan ejemplos de fraude, sino que tienen fraude o manipulación sistematizada y ocultamiento de los efectos adversos. Luego está el consumo, que en 20 años se ha duplicado. Ahora estamos en 24 recetas por habitante al año y en 2003 estábamos en 11 o 12.
¿Por qué ha sucedido esto?
Muchos medicamentos han sido comercializados con una eficacia que es más bien supuesta, basada en variables digamos vicariantes o subrogadas, como el colesterol para el infarto o la osteoporosis para las fracturas o la carga viral para enfermedades víricas, etcétera. Es conveniente estudiar cuáles son los efectos de este aumento del consumo sobre la salud pública. He intentado cuantificar cuántas enfermedades, cuántas incapacidades, cuántas muertes son causadas por medicamentos. No tanto para hacer una estimación muy precisa, que no la hay porque faltan estudios, pero sí para por lo menos dar el orden de magnitud logarítmica de la cuestión. Si son diez por millón, 100 por millón, o 10.000 por millón. En España hay más de medio millón de ingresos hospitalarios atribuibles a medicamentos; hay unas –y esto tomando las estimaciones más conservadoras– 16.000 muertes a causa de efectos indeseados de medicamentos solo en hospitales.
¿Cómo hemos llegado hasta aquí?
Nos interesa estudiar el eslabón final de la cadena de desarrollo de un fármaco, el consumo. Pero si todos los demás (investigación y desarrollo, regulación, registro, autorización, decisión de financiación por el sistema sanitario público, distribución física, prescripción médica, dispensación farmacéutica) funcionan mal, el consumo no funciona bien. Y es lo que intento mostrar, centrándome en los aspectos que yo conozco más, que no es la fabricación sino la regulación en términos de farmacovigilancia de los efectos adversos, las patentes y la manera en que las compañías farmacéuticas han modificado sus prácticas en los últimos años. Las compañías ya no son estos sitios donde un fármaco se investiga desde el inicio de la molécula hasta que sale al mercado, sino que los medicamentos se compran en Wall Street: las grandes compañías multinacionales buscan empresas biotecnológicas o de otro tipo, pequeñas, que tengan una molécula interesante y las compran enteras o les compran el producto y ya está.
¿Cómo se podría remediar o al menos mitigar esta deriva que denuncia?
Yo propongo algunos ejes referidos al sistema sanitario, porque aunque hay que cambiar muchas cosas, donde se prescriben o consumen los medicamentos, donde ocurren los efectos adversos, es en el sistema sanitario. El primero es que el sistema sanitario español debería seleccionar entre los medicamentos autorizados cuáles son los que más le conviene. Es imposible la gestión del conocimiento de los médicos sobre las 16.000 especialidades farmacéuticas que pueden prescribir, con 3.000 moléculas comercializadas como fármacos en España. Esto un médico no lo puede conocer. Cuando hay tantos fármacos diferentes en el mercado pero que en realidad muchos son esencialmente lo mismo, se crea confusión a todos los niveles de la cadena del medicamento: en el control de calidad por parte de la agencia, en la selección de los medicamentos a financiar por el sistema sanitario, entre los prescriptores. Y crea confusión en los usuarios. He hecho a lo largo de mi vida profesional estudios en los que se mostraba que una persona tomaba el mismo fármaco varias veces y en dosis muy altas porque estaba en marcas comerciales diferentes.
El segundo sería que es inaudito que los médicos del sistema sanitario público se informen sobre los medicamentos por la industria farmacéutica. La información que circula sobre medicamentos es elaborada por los departamentos de marketing de la industria farmacéutica. No puede ser.
El tercer eje, la formación continuada. El 90% de los cursos acreditados de formación continuada en España son alentados, promovidos, pagados o presentados directamente como promovidos por la industria farmacéutica. Los conocimientos para la práctica de cualquier especialidad médica se duplican cada siete u ocho años. Esto quiere decir que a lo largo de los 40 años de una vida profesional se duplicarán seis o siete veces. ¿Quién les cuenta las novedades? Básicamente, la industria. Hay mucha ósmosis entre compañeros, naturalmente, pero la industria también mete sus mensajes en esta ósmosis, que de este modo no aparecen como mensajes con un origen comercial.
El cuarto es el diálogo con los profesionales y el fomento de su participación en las políticas farmacéuticas, centrado en los problemas de cada uno de los centros de salud. En Barcelona no es lo mismo en el barrio del Raval, el más pobre, o en de San Gervasio, el más rico. Las prioridades en cada uno de estos sitios son diferentes y la manera de usar los medicamentos puede que también varíe.
El quinto sería evitar los conflictos de intereses. El primer conflicto de interés nace de la aprobación de los Presupuestos Generales del Estado en el 2015, era un gobierno de Rajoy. Ciudadanos le dio el voto favorable en casi el último momento por una enmienda transaccional por la cual los ingresos en especie recibidos por los médicos como inscripciones a congresos, pagos de viaje, hoteles y todo eso por la industria farmacéutica no tributan. Los parlamentarios de Ciudadanos y del PP decían que si no hacían esto cómo se iban a formar los médicos, que es ya reconocer de entrada que se renuncia a que el sistema sanitario tenga una formación continuada.
Igual uno es un poco ingenuo, pero esto es de lo que más me ha sorprendido del libro. Esas prácticas, que uno pensaba más de países como EEUU, en la que las farmacéuticas se llevan a los médicos a congresos o estancias para ‘venderles’ fármacos y explicarles cómo funcionan.
En el libro hablo de cuánto gasta la industria en promoción comercial y a cuánto tocaría por médico…
Dice que se gastan más en marketing que en I+D, que parece bastante indicativo de cómo funciona el sector.
En 2011 la industria dedicó un 24,4% de su volumen de negocio a promoción comercial. El volumen de negocio fue de 15.800 millones de euros en 2011, de manera que se dedicaron a promoción 3.857 millones, sin contar el negocio generado fuera del sistema sanitario público. La mayor parte de este gasto se dedica a la promoción dirigida a los médicos. Y si en 2019 había 207.000 médicos en ejercicio en España, esto da 18.600 euros de media por médico colegiado y año, algo que quizá ayude a entender cómo la industria teledirige el comportamiento prescriptivo del conjunto del Sistema Nacional de Salud. Y añado: sin que el teledirigido se dé ni siquiera cuenta.
Hablemos de cosas concretas. Por ejemplo, escribe que hay medicamentos que se toman muchísimo más de lo necesario, sea en el tiempo o en cantidad, y cita como ejemplo de esta práctica el omeprazol.
Toda la presión de protocolos y demás está dirigida a promover el uso crónico continuado y si puede ser durante toda la vida del mismo medicamento. Por eso la industria tiene poco interés en desarrollar antibióticos, porque los tratamientos antibióticos duran poco. La idea de un departamento de marketing, tal como la oí hace unos años, es que de cada visita médica se originan 35 prescripciones adicionales. ¿Cuánto cuesta una visita médica? ¿Cuántas visitas hace el visitador al día? A partir de aquí se echan cuentas. Si tú originas 35 prescripciones de un antibiótico para una semana es poco. Si originas 35 prescripciones de omeprazol es mucho mejor, porque es un cliente para toda la vida o al menos unos cuantos años.
Bajo este prisma, el omeprazol viene propuesto no como un fármaco, sino rebautizado como protector gástrico, como si fuera una tirita que le pones al estómago. Pero es un fármaco, inhibe la secreción de ácido en el estómago. Y, recuerdo, los fármacos no tienen una sola acción farmacológica. El omeprazol, aunque es extraordinariamente seguro en comparación con otros, a la larga puede producir insuficiencia renal, aumento de las fracturas de fémur, aumenta las infecciones, las diarreas graves y las neumonías. El estómago es una barrera ácida de protección de entrada de virus y bacterias, y una vez suprimida esta barrera de protección entran muchas más bacterias en nuestro cuerpo y por eso hay más infecciones. Un protector gástrico es como un bastón para caminar, por qué dejar de usarlo si va bien. Y así prosigue el consumo. En el sistema sanitario español es mucho más fácil prescribir un medicamento que retirarlo. Muy a menudo se dice que esto es porque el paciente quiere que le prescriban un medicamento. No es cierto. Los estudios indican que la gente se toma muy bien que le quiten un medicamento.
Usted también es muy crítico con el tratamiento que se hace de la depresión. ¿Por qué?
Porque los fármacos son malos. Las pruebas para decidir si un producto es antidepresivo o no son un chiste. Por ejemplo, el test de la natación forzada. Ponen a una rata en un cubo con agua y miden el tiempo que el animal está intentando escaparse hasta que para y simplemente va moviendo lentamente los brazos para mantenerse a flote. Entonces dicen que parar de luchar es como la depresión, es tirar la toalla. Si un animal dura más tiempo haciendo este esfuerzo es que el fármaco que le damos es antidepresivo. Imagínate. La cocaína haría el trabajo, o la anfetamina. Yo no les llamo antidepresivos porque es un un mal nombre. No son anti, es un nombre comercial. Los antipsicóticos no son específicos para una psicosis ni para la depresión. Los llamados antidepresivos aplanan las emociones, tanto las positivas como las negativas, y eso a algunas personas en ciertas situaciones les puede parecer beneficioso porque les quita una parte de su sufrimiento, pero no parece que haya más que eso.
Los ensayos clínicos son fraudulentos, están manipulados, publicados de manera selectiva. Los pocos en los que les ha salido el fármaco superior al placebo los han publicado varias veces, de manera que parezcan varios ensayos clínicos. También hay que ver está cómo los ensayos usan las escalas de depresión. Las escalas en patología mental o en la enfermedad de Alzheimer miden cosas que pueden no ser iguales para todo el mundo. El insomnio, por ejemplo, te cuenta hasta cuatro puntos en la escala de depresión. Querer suicidarte también te cuenta entre uno y cuatro puntos según la intensidad. Creo que no son cosas iguales. Para cada paciente, cada síntoma tiene una importancia diferente. Y después están los efectos adversos, que son muchísimos, son graves y, curioso, para varios fármacos se hacen públicos justo cuando caduca la patente del medicamento.
España es el primer país del mundo en consumo de benzodiacepinas. ¿Por qué? ¿Qué nos hace especiales?
No lo sé, pero me imagino que no será porque en España haya diez veces más ansiedad o más insomnio que en Alemania, donde el consumo de las benzodiacepinas es diez veces más bajo. ¿Hay diez veces menos insomnio, menos ansiedad? Me inclino por responder que no, que lo que hay son actitudes diez veces diferentes de los médicos que prescriben estos fármacos. Quizá en Alemania se prescriban igualmente, pero para una duración muy corta, como dicen los ensayos clínicos, y si puede ser no más que un uso puntual. En cambio, aquí los estudios de utilización de estos medicamentos nos indican que la enorme mayoría de los consumidores las toma desde hace más de un año y más de dos. En España es muy fácil prescribir y muy difícil desprescribir –una palabra que no me gusta nada– o retirar el medicamento.
Más cosas concretas. Le dedica un capítulo entero al colesterol, que relaciona con las enfermedades inventadas.
Inventadas o exageradas. El colesterol es un paradigma, de la invención de problemas, pero también están los analgésicos, los psicofármacos, hipnosedantes, para dormir y los medicamentos para la depresión.
Lo mismo con el colesterol, que se inventa como enemigo público número uno de la salud pública. Llevan 70 años martilleando con el colesterol, como si anunciara un infarto de miocardio y de ictus. En el libro hablo de las campañas de Mundifarma y de determinadas sociedades científicas que, pagadas por la industria, hacen campañas para insensibilizarnos contra el dolor crónico. Y vienen con estas encuestas de que un 30% o un 40% de la población padece dolor crónico que está mal tratado. ¿Qué quiere decir dolor crónico? ¿Qué es una persona que tiene dolor desde que se levanta hasta que se va a la cama? ¿Tanto si se mueve como si está quieto? Hay muy pocas que tengan un dolor continuado que no se marcha ni cambiando de posición ni a ninguna hora del día. Pero se promueven analgésicos para todo el día, y así se favorecen las dependencias de los opiáceos y demás.
Lo mismo ocurre con el insomnio. Se receten fácilmente medicamentos para dormir, pero de unas semanas cómo ha ido, si se lo ha tenido que tomar cada día o no, etcétera. Los sistemas sanitarios de acceso universal fue una gran conquista generalizada en Europa después de la Segunda Guerra Mundial. Trataban a personas enfermas, para restablecer la salud, pero ahora trata mayoritariamente a personas sanas haciéndolas creer que están enfermas. ¿Cuántas mujeres que sufren malos tratos de género reciben antidepresivos sin que el médico haya aclarado a qué se debe aquella depresión, aquella ansiedad y aquel malestar? No lo sabemos, pero muchísimas.
Antes ha comentado que a la industria no le interesa desarrollar antibióticos porque son tratamientos muy cortos, y eso me ha llevado a pensar en una conversación que tuve el otro día con un colega suyo, Damián García Olmo, sobre terapias avanzadas. Me dijo que se está viendo que una de las cosas que quizá puedan hacer en algún momento es cambiar un medicamento crónico por un solo pinchazo. Estas dos cuestiones son contradictorias. ¿Van a permitir las farmacéuticas que les quiten negocio en forma de medicamentos crónicos?
Hay un capítulo en el libro que tiene un subtítulo que dice que curar no es negocio. Hace unos diez años se sacaron unos medicamentos para la hepatitis C, el Sovaldi o Sofosbuvir y demás, los primeros de la nueva serie de medicamentos ultra caros, con un coste de ochenta y tantos mil dólares por paciente. La empresa que los sacó, la multinacional estadounidense Gilead, se hizo muy rica, sus acciones subieron mucho. Pero la gente con hepatitis C o bien son pobres, que no llegarán nunca al tratamiento, o bien son ricos que ya se curaron. Las previsiones de ingresos caían al cabo de pocos años y un informe de Goldman Sachs se preguntaba si curar a los pacientes es un modelo comercial sostenible. “Un modelo comercial sostenible”. Esta frase resume la contradicción entre salud y mercado. Cuando a uno lo curas ya no es cliente, y lo que busca la industria son clientes.
¿Cuál cree que debe ser el papel del sistema público en todo este proceso? En el libro menciona el caso específico del dolor de espalda. Las ‘farmas’, dice, no lo van a estudiar porque no les interesa. Debería hacerlo el sistema sanitario público. ¿Es viable que el que el sistema público decida sobre qué vamos a investigar y qué vamos a desarrollar (y en consecuencia qué no)?
Es viable porque el sistema público es muy potente si quiere, pero depende de las prioridades que tenga y la comprobación de la efectividad no está entre las prioridades actuales de los sistemas sanitarios públicos. En el libro cuento varios ejemplos de cómo los sistemas sanitarios públicos han podido o bien reconfirmar la eficacia de algún fármaco o bien descartar que un fármaco más caro era mejor que otro más barato y más seguro para la misma enfermedad. La investigación en el sistema sanitario público tiene, si es independiente, la ventaja de la independencia y la transparencia. Otra ventaja es una comprobación de lo que en la práctica se ha visto en ensayos clínicos, la diferencia entre lo que en términos técnicos llamamos eficacia y efectividad.
La eficacia es la capacidad de un fármaco o de cualquier intervención para mejorar el curso de una enfermedad y se mide en comparación con placebo en un ensayo clínico. La efectividad es cómo esto se traduce en la práctica, y no siempre son lo mismo. La tercera ventaja sería la investigación en el marco del sistema de salud, los ensayos clínicos y demás estudios cuando no se mete con calzador, cuando la investigación sale de preguntas que se hacen los propios médicos para mejorar su práctica y ellos participan en la investigación. Esto aumenta de una manera extraordinaria el caudal de conocimiento del sistema de salud.
Porque lo que es indubitado es que el sistema público en muchas ocasiones pagamos dos veces, y a un alto coste, por lo mismo. Primero financia la investigación básica, luego llega un momento en el que se le vende ese producto a una gran farmacéutica y cuando esta lo desarrolla y lo pone en la calle lo volvemos a comprar a precio de mercado.
Efectivamente pagamos dos veces y además la segunda vez que pagamos pagamos lo hacemos por unos datos que han producido las compañías farmacéuticas. Ellas son las que realizan los ensayos clínicos sin controles externos. Los datos no son accesibles para investigadores interesados y esto a mí me parece realmente un problema.
Este texto es absolutamente demoledor en los datos que aporta y en sus reflexiones. Consideramos que su lectura proporciona claves para entender la situación actual de acceso a los tratamientos oncológicos en términos de coste y seguridad clínica.
Sus conclusiones finales expresando que “La realidad del poder y el dinero que se manifiesta a través de la política contemporánea significa que SACT continuará siendo dominado y dirigido por intereses privados hasta que una crisis obligue a un cambio radical. Esperar a que las instituciones críticas de la sociedad reflejen honestamente la verdadera magnitud de los beneficios, así como la amplia brecha de asequibilidad, bien podría ser similar a esperar a que el infierno se congele” son tristes, pero al a vez señalan la posibilidad de una esperanza
Es de un valor singular y recomendamos vivamente su lectura.
Resumen
Los medicamentos contra el cáncer se han convertido en una de las tecnologías médicas mundiales más dominantes. Generan enormes ganancias para la industria biofarmacéutica, así como alimentan las actividades de investigación y desarrollo de los financiadores públicos, las organizaciones de pacientes, las comunidades clínicas y científicas y ecosistemas políticos federales enteros.
El desajuste entre el precio, la asequibilidad y el valor de muchos medicamentos contra el cáncer y la necesidad global ha generado un debate político significativo. Si. embargo, vemos pocos cambios en los comportamientos de cualquiera de los principales actores, desde los financiadores públicos de la investigación hasta las autoridades reguladoras.
En este análisis de políticas examinamos si, teniendo en cuenta el dinero y el poder inherentes a este sistema, se puede lograr algún consenso sobre una política global racional para ofrecer medicamentos contra el cáncer asequibles y equitativos que ofrezcan consistentemente beneficios clínicamente significativos.
Un poco de historia
En 2012, Scannell et al [1] publicaron un artículo fundamental que se convertiría en lectura obligatoria en el sector biofarmacéutico. En su corazón estaba un diagnóstico de por qué la productividad de la industria estaba disminuyendo. Para expresar su idea acuñó el término Ley de Eroom (ley de Moore, al revés). Corresponde con su observación de que el descubrimiento de medicamentos se estaba volviendo más lento y más caro con el tiempo, a pesar de las mejoras en la tecnología; una tendencia observada por primera vez en la década de 1980. El costo ajustado a la inflación del desarrollo de un nuevo medicamento se duplicaba aproximadamente cada nueve años. Pero en la década de 1990, esta tendencia iba a revertirse drásticamente gracias al comienzo de la era dirigida molecularmente en los medicamentos contra el cáncer. De la noche a la mañana, los biofarmacéuticos de oncología pasaron de estar en un remanso a ser los salvadores del sector en su conjunto.
Como Scannell señaló más tarde en una entrevista en 2020, «los rendimientos de la I+D son estocásticos y sesgados. La industria obtiene una cantidad desproporcionada de sus beneficios con algunas drogas muy importantes. Incluso la economía de las grandes empresas es muy sensible a uno o dos productos, y mientras algunas personas en la industria estén ganando, es muy difícil para otras personas entrar en el juego. Por lo tanto, si algunas empresas de la industria parecen estar bien, las empresas pueden expresar de manera plausible su confianza en sus canales de producción y en los científicos, y defender el uso del dinero de los accionistas, a pesar de que pudieran perder parte de él».
¿Por qué es tan importante esta pieza de la historia? Porque puso en marcha una serie de tendencias globales que nos llevan a donde estamos hoy. Y explica por qué, a pesar de la abrumadora evidencia de que los ecosistemas actuales que rigen la investigación y el acceso al mercado de los medicamentos contra el cáncer sistémicos (SACT) no son asequibles, equitativos o sostenibles, poco parezca estar cambiando. Diez años después de la primera Comisión de Oncología de Lancet sobre la atención del cáncer asequible en países de altos ingresos [2], todos los países están luchando por abordar un modelo sostenible para una SACT asequible, así como los costos de las oleadas de tecnologías nuevas y costosas en radioterapia, cirugía, imágenes, etc.
Los biofármacos contra el cáncer: se trata fundamentalmente de ganar dinero
Este titular de declaración molesta a la gente. Les molesta porque no nos gusta enfrentarnos a la realidad. Y esa realidad es que la industria biofarmacéutica trata de obtener un beneficio para los accionistas. Y el beneficio, en sí mismo, no tiene un límite. El motor de esto es EE. UU., que es, a nivel mundial, el principal actor cuando se trata de incentivar la I+D biofarmacéutica oncológica. Y es el ecosistema legislativo de EE. UU. el que permite a estas empresas un poder de fijación de precios infinito. Un poder que tiene implicaciones globales. Dicho sin rodeos, el sistema de I+D sobre el cáncer está demasiado incentivado y esto conduce a medicamentos marginales.
Un análisis más detallado sobre los costos de I+D y los rendimientos de la inversión ilumina lo fuertes que son las corrientes dirigidas al beneficio cuando se trata de oncología. Vale la pena señalar que en este análisis no se incluyó ninguno de los medicamentos de inmunooncología más nuevos porque es demasiado pronto para evaluar los costos de su I+D y ROI. Sin embargo, el análisis incluyó el tramo más reciente de medicamentos contra el cáncer personalizados basándose en datos financieros detallados presentados ante la Comisión de Bolsa y Valores de los Estados Unidos (SEC) [3].
A lo largo de 23 años de I+D de medicina contra el cáncer, el costo medio en dólares de desarrollar un medicamento contra el cáncer por las empresas farmacéuticas ha sido de alrededor de 4.400 millones de dólares. Pero esto oculta una enorme gama de costos, desde «solo» 276 millones de dólares para Dinutuximab para la terapia posterior a la consolidación para el neuroblastoma de alto riesgo infantil, hasta 13,4 y 15.800 millones de dólares, respectivamente, para Durvalumab (utilizado para tratar ciertos tipos de cáncer de vejiga, pulmón y tracto biliar) e Isatuximab (utilizado en el tratamiento del mieloma múltiple de cáncer de sangre).
Esto refleja el hecho de que los verdaderos costos de la I+D cubren una gama muy amplia, y estos gastos no tienen y nunca han tenido ninguna relación con los precios posteriores en ningún mercado. Además, ninguna de estas cifras tiene en cuenta la financiación pública de I+D que está íntimamente entrelazada en el sector privado [4]. El precio de SACT ha atraído grandes cantidades de capital de I+D, lo que significa una competencia cada vez mayor en la reducción de los espacios terapéuticos, la duplicación y los bajos rendimientos sociales.
Los datos sobre el rendimiento de las inversiones son aún más sorprendentes. El primer punto para considerar es que llevar cualquier medicamento contra el cáncer al mercado genera rendimientos sustanciales para la(s) empresa(s), independientemente del retorno de la inversión. Incluso llevar un medicamento contra el cáncer a lo largo del proceso garantiza flujos de capital sustanciales en términos de inversión interna en I+D.
En general, el retorno de la inversión de los medicamentos contra el cáncer con una madurez suficiente, es decir, lanzados entre 1997 y 2015, está entre el 435 y el 551%. Una vez más, esto esconde una gran variación. Un número sustancial de medicamentos contra el cáncer hasta la fecha tienen un retorno de la inversión negativo o fijo tan bajo de menos 78-87 % en algunos casos. Sin embargo, algunos medicamentos contra el cáncer lanzados a finales de la década de 1990 hasta mediados de 2000 han generado ROIs astronómicos; por ejemplo, Erlotinib (2794%) (cáncer de páncreas y de pulmón), Trastuzumab (3421%) (cáncer de mama), Rituximab (2523%) (Linfoma no Hodgkin) y Bevacizumab (3200%) (colon, pulmón, glioblastoma y cánceres de células renales). Esto refleja el hecho de que el modelo de negocio de oncología todavía está impulsado por éxitos de taquilla.
Si bien este trabajo proporciona más claridad basada en cifras reales presentadas ante la SEC de EE. UU., también arroja luz sobre la vasta complejidad financiera de la industria biofarmacéutica. De hecho, sabemos que el retorno de la inversión es casi seguro más alto que las cifras calculadas y que estas cifras continúan aumentando y expandiéndose año tras año. Hay evidencia convincente de que es probable que el desarrollo de medicamentos contra el cáncer impulsado por el diagnóstico de los medicamentos de precisión reduzca los costos de I+D. Sin embargo, no hay evidencia de que esto fluya hacia precios más bajos aguas abajo. En todo caso, los precios premium, «lo que el mercado soportará», están subiendo los precios y los costos de los medicamentos contra el cáncer en todos los ecosistemas y esto continuará.
Sabemos por el trabajo de Lazonick [5] y otros que, contrariamente a la narrativa predominante, la mayoría de las ganancias de la industria farmacéutica no se devuelven a la I+D, sino que se utilizan para la recompra de acciones en el mercado abierto. Uno de los principales mecanismos para mantener los valores altos. Estas recompras de acciones, que se suman a los pagos de dividendos, se realizan en nombre de «maximizar el valor de los accionistas» (MSV). Las recompras dan impulsos manipuladores al precio de las acciones de la empresa y, al reducir el número de acciones en circulación, aumentan las ganancias por acción (EPS), un indicador ampliamente aceptado del rendimiento de una empresa. El aumento de los precios de las acciones enriquece a los altos ejecutivos, que reciben la mayor parte de su compensación en forma de opciones sobre acciones y premios de acciones [6].
¿Por qué hay tal impulso para impulsar nuevos medicamentos contra el cáncer que tienen perfiles de beneficios clínicamente significativos bajos o inexistentes? Hasta cierto punto, la respuesta se basa en el poder de la incentivación para impulsar el capital tanto en el sector biotecnológico como en el farmacéutico. A través de tal especulación, es muy posible que los inversores obtengan enormes recompensas al operar con acciones biotecnológicas y farmacéuticas, independientemente del beneficio que ofrezcan.
Pisano [7] argumentó que «solo aproximadamente el 20 % de todas las empresas que existen hoy en día tienen algún producto en el mercado o están ganando regalías basadas en productos comercializados por socios. Por lo tanto, la gran mayoría de las empresas de biotecnología que trabajan en oncología son esencialmente entidades de I+D». Así que, el problema es que toda la estructura y el marco de incentivos que rigen la biotecnología y la industria farmacéutica están orientados a un tipo específico de comportamiento. A falta de reformar completamente todo el mercado industrial de capital, comenzando en los EE. UU., esperar que la industria se comporte de manera diferente a lo que está haciendo en este momento es un callejón sin salida. Es un callejón sin salida ideológico y técnico. Discutir sobre los derechos y los errores de las ganancias de la industria en oncología no tiene sentido. El sistema está orientado a la maximización de los beneficios de forma completamente independiente a los costos de I+D y es insensible a si los medicamentos ofrecen un beneficio terapéutico significativo o si los medicamentos contra el cáncer tienen un precio «justo» para cualquier país determinado.
La captura privada del público
SACT domina la I+D de oncología global. El sector privado ha capturado todas las principales vías del desarrollo preclínico y clínico de medicamentos contra el cáncer, incluidos los ECA [8]. Al hacerlo, el sector privado está ahora en una posición de liderazgo para establecer la agenda de investigación, así como el diseño de los estudios clínicos. En ese sentido, no es de extrañar que los puntos finales que aseguran el camino más rápido posible a través del desarrollo clínico al mercado ya sean válidos o no, hayan dominado. La manipulación de los datos se ha normalizado dentro del ecosistema de medicamentos contra el cáncer; exagerando la eficacia y minimizando la toxicidad y los problemas de tolerabilidad. En el primero, la cuestión de los criterios de valoración no validados o parcialmente validados se ha expresado en voz alta, sin embargo, esto ha tenido poco impacto en el diseño de los ensayos clínicos ni en la aprobación regulatoria [9, 10]. El blanqueo de la toxicidad grave también se ha convertido en una norma. El desarrollo del inhibidor de mTor everolimus es un caso particularmente atroz de minimizar la toxicidad [11]. Un progreso significativo en el desarrollo de medicamentos contra el cáncer requiere, como han dicho Labadie y Fojo [12], «interpretación rigurosa de los datos y diseños equilibrados de ensayos clínicos. También requiere estrategias que limiten la censura informativa en la evaluación de resultados a un máximo de un poco por ciento para que no los declaremos ininterpretables». Pero estos fracasos sistémicos se alejan con la noción de que se pueden llenar tales lagunas probatorias. La suposición ha sido que no importa que algunos SACT entren en el ecosistema clínico de forma temprano¡a con una incertidumbre tan enorme en cuanto a su valor clínico y económico porque la investigación posterior a la comercialización separará el trigo de la paja. Aquí es, supuestamente, donde la Evidencia del Mundo Real (RWE) viene al rescate. La realidad es algo diferente, con déficits impactantes en la calidad y el rango de RWE que hacen que la mayoría de los estudios no sean aptos para guiar la práctica o la política clínica [13, 14].
Las consecuencias de un ecosistema de I+D construido en torno al sector privado encuentran una causa común en la cultura del beneficio. Gran parte de esto está impulsado por la cultura de I+D de EE. UU. Como dijo Sahlins [15], «la organización sistemática de la universidad de investigación por la subjetividad comercial y la actividad empresarial o, en otras palabras, la búsqueda del conocimiento desinteresado por parte de personas interesadas». No hay contradicción entre el beneficio privado y el bien público en este modelo. Las instituciones superiores (universidades y los principales centros oncológicos) actúan como subcontratistas de la economía de la biociencia. Las instituciones públicas y todo el dinero público sirven a la sociedad al servir también al sector privado, al que la sociedad debe su riqueza, conocimiento y forma de vida. En casi todos los análisis de las publicaciones de investigación, SACT (y los dominios de investigación asociados) dominan los resultados de la investigación, ya sea si uno mira el ecosistema de I+D oncológica de EE. UU. o los sistemas más socializados en toda Europa [16]. Las principales organizaciones públicas de financiación de la investigación también participan en esta captura. Sus carteras de investigación estaban muy orientadas hacia la I+D biofarmacéutica alineada por el sector privado. Esta dominación es total. Y los países de altos ingresos no están solos. Las principales economías emergentes, como China, también están muy invertidas en SACT, particularmente en productos biológicos / inmunooncología de próxima generación [17]. Los sistemas nacientes de I+D, como los de MENA [18] y el África Subsahariana [19], también están encontrando una causa común en los mismos dominios biofarmacéuticos. Esto plantea preguntas sobre lo que ahora constituye una verdadera investigación oncológica del bien público. Las estrategias de las organizaciones de financiación de la investigación pública se han convertido, esencialmente, en farmacéuticas [20]. Esto no es solo el beneficio del sector privado, que en sí mismo es completamente normativo, sino que también se trata de la dependencia y la adicción de los países, los grupos de defensa de los pacientes, los financiadores públicos de la investigación, las universidades, así como las comunidades clínicas y científicas, al flujo sustancial de financiación del sector privado que viene con la alineación estratégica con las industrias biofarmacéuticas.
¿A alguien le importa de verdad?
Empecemos con el beneficio. De 2000 a 2018, 35 grandes compañías farmacéuticas reportaron ingresos acumulados de 11,5 billones de dólares con un beneficio bruto de 8,6 billones de dólares [21]. Estos son asombrosos y la oncología es uno de los principales contribuyentes a estas cifras [22]. Si bien ha habido una buena cantidad de diálogo en torno a las obligaciones especiales de la industria farmacéutica durante las emergencias, por ejemplo, las pandemias cuando se trata de fijar los términos del debate en torno a los medicamentos contra el cáncer han sido escasas y desiguales [23]. En general, los argumentos de que la industria tiene que equilibrar sus responsabilidades hacia los pacientes con las expectativas de ganancias de sus partes interesadas han encontrado poco terreno fértil donde la rentabilidad de las grandes compañías farmacéuticas es significativa [21]. En general, ningún financiador público de la investigación, el gobierno, las organizaciones de pacientes con cáncer o las organizaciones profesionales como ASCO o ESMO han pedido nunca una restricción de beneficios, o incluso una fijación de precios proporcional (es decir, justo).
El consenso es que la fijación de precios de SACT en la mayoría de los países sin una gobernanza significativa es injusto y el impacto en los pacientes es, en la mayoría de los sistemas, ruinoso [24]. Superan los umbrales de asequibilidad y relación calidad-precio y no están justificados por los costos de investigación y desarrollo [25]. Para ser justos con la industria, en muchos países también hay un comportamiento asombroso en la búsqueda de colaboraciones entre el fabricante y el paciente. En muchos países, múltiples actores, incluidos hospitales y médicos, plantean su «papel » a lo largo de la cadena de suministro [26]. Hay muy poca discusión sobre esto. Pero lo que esto significa es que el conjunto de problemas es sistémico. Si te importan los precios inasequibles, entonces no es suficiente simplemente abordar la industria, debes abordar toda la cadena de ganancias.
Parte del problema de los precios también se debe a la I+D «desperdiciada». Por ejemplo, los fracasos de los programas en torno al desarrollo de inhibidores específicos del receptor del factor de crecimiento 1 similar a la insulina (IGF-1R) pueden haber costado alrededor de 1.600 millones de dólares. Tales fallos reflejan un desperdicio evitable en términos de tiempo, dinero y pacientes. Por una variedad de razones, estos «viajes al fracaso» no dejan ninguna lección aprendida [27]. Y el sistema está orientado a repetir estos costosos errores. Aguas abajo, estos fallos, y los costos, se convierten en precios más altos. Más allá de estas pérdidas por tuberías, el sistema también está cada vez más orientado a permitir el acceso al mercado a nuevos SACT (o nuevas indicaciones) que, no ofrecen un beneficio clínicamente significativo. Sin embargo, nuestros sistemas regulatorios son tan promiscuos y tan conflictivas son las comunidades que defienden todos los nuevos SACT (o indicaciones) de tal forma que lo que vale la pena y lo inútil se mezclan con los mismos aplausos. En general, nadie plantea ninguna objeción. Las organizaciones de pacientes, las organizaciones públicas de financiación de la investigación, los organismos profesionales, rara vez plantean un toque de crítica. Y, por supuesto, el problema entonces es que los fondos públicos se gastan tanto en un nuevo SACT significativo y que vale la pena como, otro francamente, sin valor. Y esto tiene seria capacidad de producir costos. Hay algunos nuevos tipos sobresalientes de SACT (y nuevas indicaciones), pero estos están llenos de una gran cantidad de cosas que no ofrecen un beneficio clínicamente significativo.
Por qué las respectivas comunidades de cáncer parecen preocuparse tan poco por esto es, hasta cierto punto, una medida de lo profundos que son los conflictos de intereses. Hay una asombrosa dependencia del dólar farmacéutico en casi todas las evaluaciones de la oncología. Las organizaciones de pacientes existen debido a la financiación del sector privado, las carreras científicas y clínicas se hacen aprovechando las grandes subvenciones farmacéuticas, los gastos generales de estas subvenciones apuntalan los sistemas universitarios y hospitalarios, la participación en ensayos farmacéuticos novedosos o a mayor escala aporta poder y estima, y los organismos profesionales pueden expandirse y prosperar por completo debido a la sustancial financiación del sector privado. Es difícil preocuparse por las consecuencias posteriores de la mala I+D y las débiles regulaciones sobre los precios cuando estás ganando tanto dinero. En países como los EE. UU., los pagos directos a oncólogos médicos que establecen directrices nacionales son ahora la norma [28]. EE. UU. no está solo con importantes problemas de transparencia, incluso en países socializados como el Reino Unido [29]. Los aumentos significativos en los pagos ad hominen ocultan un problema mayor. El hecho es que hay flujos de financiación de varios millones en hospitales y universidades con poco o ningún escrutinio en cuanto a si los ensayos o la investigación reflejan una pregunta importante o un estudio bien diseñado. De hecho, el avance profesional y la estima se deben casi en su totalidad a la cantidad de dinero que uno aporta, no a si la investigación en cuestión tiene algún valor [30].
En última instancia, es para que las autoridades reguladoras – FDA, EMA, etc. – actúen como árbitros del bien público en lo que respecta a los medicamentos. Pero a juzgar por el criterio en cuanto a si las nuevas SACT o las nuevas indicaciones están proporcionando consistentemente un beneficio clínicamente significativo, la regulación ha fracasado. Se considera que la mitad de los ECA que se utilizaron para apoyar la aprobación de la EMA entre 2014 y 2016 tenían un alto riesgo de sesgo en función de su diseño, conducta o análisis. Y las publicaciones de revistas posteriores no reflejaron las limitaciones clave de la evidencia disponible identificada en los documentos regulatorios [31]. Demasiados nuevos SACT o nuevas indicaciones obtienen la aprobación regulatoria sin una demostración clara de su beneficio (mejorando la supervivencia general y/o la calidad de vida) [32]. Solo un tercio de las aprobaciones de la FDA de agentes terapéuticos para su uso en oncología de tumores sólidos para el período 2017-2021 se consideró que ofrecían un beneficio clínicamente significativo [33]. Pero, aparte de una serie de artículos de opinión y publicaciones analíticas, este extraordinario desequilibrio no ha generado ni una sola onda de respuesta política. Los comportamientos de las autoridades reguladoras permanecen intactos, y las autorizaciones, en todo caso, han cobrado ritmo a medida que el mantra de «rápido en el mercado» se convierte en el estándar político para ofrecer beneficios al paciente [34, 35].
Poder y dinero
La causa principal del problema de la SACT está directamente en los EE. UU. Su incentivo excesivo de la I+D biofarmacéutica contra el cáncer, su capacidad para proporcionar a la industria un poder infinito cuando se trata de establecer precios, los sistemas públicos y privados completamente entrelazados (gracias a la Ley Bayh-Dole de 1980) y el enorme impacto global de la FDA a través de iniciativas como el Proyecto Orbis han establecido la agenda de poder y dinero.
En la búsqueda de encontrar soluciones, hemos sido testigos de una verdadera cornucopia de publicaciones e iniciativas en la última década que buscan iluminar y abordar el conjunto de problemas entrelazados. Se han creado movimientos que buscan restablecer el equilibrio. El comité de la Lista de Medicamentos Esenciales para el Cáncer de la OMS ha abordado los precios y la accesibilidad como aspectos clave de sus deliberaciones [36], las iniciativas de Choosing Wisely han tenido como objetivo los precios excesivos [37], iniciativas nacionales como la Red Nacional del Cáncer de la India han tratado de establecer precios más justos a través de adquisiciones conjuntas [38], y se han establecido movimientos como Common Sense Oncology [39] para tratar de promover más ampliamente los problemas en el diseño deficiente de ensayos. En ese sentido, hay más conciencia y sensibilización en torno a los problemas que nunca. Algunas organizaciones globales, Clinton Health Access Initiative, por ejemplo, han defendido prácticamente un acceso mejor y más asequible a los medicamentos contra el cáncer. Algunos países también han trabajado duro para tratar de gestionar su ecosistema SACT tanto en el lado de la demanda como en el lado de la oferta. Y en comparación con hace 20 años, estamos inmensamente mejor en términos de SACT más efectivo para toda una gama de cánceres.
Pero para un momento y pregunta qué ha cambiado realmente. ¿Ha mejorado la narrativa? ¿Tenemos discusiones realistas más equilibradas? ¿Son los precios más justos y, a nivel mundial, los regímenes SACT son ahora más asequibles? Ni un poco. En todo caso, el bombo y la hipérbole acaban de empeorar [40]. El poder inherente que aporta tanto dinero ha capturado franjas de territorio; financiadores públicos de la investigación, autoridades reguladoras e incluso regiones enteras [41]. Podemos escribir sobre cómo los determinantes comerciales están desequilibrando los servicios y sistemas hasta que la tristeza domine nuestra cara [42], pero, al igual que el coro griego, las palabras no tienen ningún impacto en los actores principales. No hay una pizca de evidencia de que, tomado en su conjunto, el ecosistema SACT esté cambiando de rumbo. Los canales de oncología continúan expandiéndose, las autoridades reguladoras continúan reduciendo los requisitos probatorios para la entrada en el mercado, los organismos de pacientes y profesionales continúan animando en voz alta a todos y cada uno de los nuevos medicamentos contra el cáncer, independientemente del precio o del beneficio clínico real, y los responsables de la formulación de políticas se quedan aún más atrás en su capacidad para ofrecer un sistema de gobernanza. Y cuando las cosas se ponen difíciles, alguna cábala aleatoria de grupos de interés e industria utiliza su dinero y poder para jugar a la política, y entonces los gobiernos ceden de forma habitual. Uno de los ejemplos más atroces de esto es la creación del Fondo de Medicamentos contra el Cáncer por parte del Reino Unido [43].
Una especie de conclusión
Uno se enfrenta a dos opciones. Acepta que el progreso es un vicio privado con beneficio público. Esencialmente alinearse con la posición de Bernard Mandeville en Fable of the Bees (1705) de que la codicia viciosa, debidamente canalizada por una política hábil, conducirá a una cooperación invisible y al beneficio público. Esposar a cualquier virtud superior es mera hipocresía. En este mundo, el único desafío es externo. Si, por ejemplo, China tuviera que ejercer su considerable músculo biofarmacéutico en oncología para socavar masivamente los precios mundiales. La otra opción está más en línea con la idea de Rawlsian de justicia social. En este mundo se construye un nuevo contrato social que realmente refleja el valor equitativo. Todas las instituciones respectivas se alinean a lo largo de esta columna vertebral común del bien público. Los precios realmente reflejan el beneficio clínico y se establecen en un nivel justo que maximiza el acceso de los pacientes. La I+D se incentiva en el valor social y no en el beneficio.
Para este autor, esta última posición es emocionalmente la más gratificante. Pero, ¿es realpolitik? La realidad del poder y el dinero que se manifiesta a través de la política contemporánea significa que SACT continuará siendo dominado y dirigido por intereses privados hasta que una crisis obligue a un cambio radical. Esperar a que las instituciones críticas de la sociedad reflejen honestamente la verdadera magnitud de los beneficios, así como la amplia brecha de asequibilidad, bien podría ser similar a esperar a que el infierno se congele [44]. Mientras sigamos siendo adictos al dinero y al prestigio, nada cambiará. Pero las ideas a lo largo de la historia siempre han tenido el poder de mover lo inamovible. Y las revoluciones pueden venir de los sectores más improbables. Lo que finalmente puede descarrilar la situación que tenemos hoy puede no tener nada que ver con un cambio directo de la política y la práctica de la oncología. Es más que probable que sea, como discute el historiador Niall Ferguson en la Gran Degeneración (2013), una confluencia de externalidades que simplemente provoquen que las personas y los países que financian el sistema de la manera en que lo hacemos ahora pagando altos precios por un mal valor cambien de rumbo.
Atención lector/a
Las referencias bibliográficas del artículo se encuentran disponibles en la edición original.
Hemos elegido poner a disposición de nuestras y nuestros lectores este editorial de The Lancet por su claridad y contundencia. Durante este mes la cantidad de artículos publicados exponiendo la trayectoria del Acuerdo ha sido muy notable. La lectora o lector interesado podrá en el apartado de Informes y Documentos acceder a algunos de ellos.
Sin embargo creemos que como señala esta espléndida Editorial esta oportunidad de conseguir un acuerdo real y efectivo que garantice la prevención, preparación y respuesta a pandemias futuras protegiendo a todos los seres humanos está en riesgo.
El Órgano Intergubernamental de Negociación (INB), que tiene la tarea encargada por la OMS de elaborar un instrumento internacional sobre prevención, preparación y respuesta a la pandemia, se sentará por novena y última vez del 18 al 29 de marzo. En los 2 años desde que se reunió por primera vez, se han gastado cientos de horas y costos desconocidos, pero el ímpetu político ha muerto. La convención se encuentra ahora en una coyuntura crítica: el texto final para que los países ratifiquen se presentará en la Asamblea Mundial de la Salud en mayo. Con solo días limitados de negociación y un largo camino por recorrer para asegurar un acuerdo significativo, es ahora o nunca para un tratado que puede hacer del mundo un lugar más seguro.
Es difícil de recordar a veces, entre las negociaciones altamente diplomáticas y técnicas, que lo que este tratado está tratando de hacer: es proteger del daño a todas las personas, en todos los países, sin importar cuán ricas o pobres. Mientras The Lancet salió a la prensa, se esperaba un nuevo borrador de texto disponible públicamente, pero a juzgar por la versión más reciente disponible, de octubre de 2023, el tratado fracasará en este objetivo. Gran parte del lenguaje está muy debilitado por la ambición inicial, llena de lugares comunes, advertencias y el término «cuando sea apropiado». Una recomendación clave del Grupo Independiente para la Preparación y Respuesta a la Pandemia, que encontró un amplio apoyo, fue la necesidad de un tratado que » abordara las lagunas en la respuesta internacional, aclarara las responsabilidades entre los Estados y las organizaciones internacionales, y estableciera y reforzara las obligaciones y normas legales». En el centro de esta recomendación estaba la necesidad de garantizar que los países de altos ingresos y las empresas privadas se comporten de manera justa, que no almacenen millones de dosis de exceso de vacuna o se nieguen a compartir conocimientos y productos que salvan vidas, y que existan mecanismos para garantizar que los países trabajen juntos en lugar de uno contra el otro. Estas cuestiones todavía representan los amplios puntos de fricción en las negociaciones actuales: el acceso y el reparto de beneficios (quién obtiene qué, cuánto y cuándo) y la gobernanza y la rendición de cuentas (en qué medida los países están destinados a hacer algo).
La palabra equidad aparece nueve veces en el texto de negociación de octubre, incluso como principio rector de todo el tratado. Pero en realidad, el artículo 12 estipula que la OMS solo tendría acceso al 20 % de los «productos relacionados con la pandemia para su distribución en función de los riesgos y necesidades de salud pública». El otro 80 %, ya sean vacunas, tratamientos o diagnósticos, sería presa de la lucha internacional ya vista durante la COVID-19 que permitió la venta de tecnologías vitales para la salud al mejor postor. La mayoría de los pueblos del mundo viven en países que podrían no poder pagar estos productos, pero el 20 % parece ser todo lo que los países de altos ingresos estaban dispuestos a aceptar. Esto no solo es vergonzoso, injusto e injusto, sino que también es ignorante. Crear y aceptar un conjunto de términos fuertes y verdaderamente equitativos sobre el acceso y el intercambio de beneficios no es un acto de bondad o caridad. Es un acto de ciencia, un acto de seguridad y un acto de interés propio. Todavía hay tiempo para corregir este juicio erróneo.
Incluso los compromisos anémicos del acuerdo están en peligro. El seguimiento independiente de si los países están cumpliendo con sus compromisos es esencial para la eficacia y la longevidad del tratado. Sin embargo, como han señalado personas como Nina Schwalbe y sus colegas, todos los indicios sugieren que los mecanismos de gobernanza y rendición de cuentas del tratado se están socavando aún más. Hay poco en el camino de obligaciones claras y exigibles para prevenir los brotes de enfermedades zoonóticas, implementar los principios de One Health, fortalecer los sistemas de salud o contrarrestar la desinformación. Es posible que los jefes de estado y el INB no vean la gobernanza de la pandemia como una prioridad ahora, pero es fundamental para el éxito de cualquier acuerdo.
Crear una convención global aceptable para todos es, sin duda, un desafío. Los objetivos de un tratado pandémico son fáciles de articular, pero muchos son difíciles de promulgar y aceptar. Puede que el INB esté haciendo todo lo posible, pero en última instancia son los políticos de los países del G7 los que deben dejar de lado los intereses creados de la industria y finalmente entender que en una pandemia no es posible proteger solo a sus propios ciudadanos: la salud de uno depende de la salud de todos. Millones de vidas que podrían haberse salvado durante la pandemia de COVID-19 no lo fueron. Lejos de hacer las paces, un puñado de países poderosos están saboteando la mejor oportunidad de traducir las lecciones de la pandemia de COVID-19 en compromisos legalmente vinculantes que nos protegerán a todos. El tratado es una oportunidad que no debe desperdiciarse.
Comparte en tus Redes
Uso de cookies
Este sitio web utiliza cookies para que usted tenga la mejor experiencia de usuario. Más info