Revista Nº 32 Septiembre 2024
AUTOR: AAJM

La importancia de este proyecto de Real Decreto para el futuro de la política farmacéutica en nuestro medio ha llevado a la Asociación para un Acceso Justo al Medicamento ( AAJM) a. realizar un análisis de su contenido y a continuación a plantear una serie de modificaciones y propuestas.
Consideramos, como así lo han puesto de manifiesto diferentes expertos, que el texto inicial presenta algunas dudas, lagunas y dificultades. A continuación, se exponen las modificaciones propuestas.
EXPOSICIÓN DE MOTIVOS
En la exposición de motivos se menciona al artículo 92.1 del RD Legislativo 1/2015 y en concreto los apartados b), c) y f) del mismo, que con posterioridad se incorporan en el texto que se propone (artículos 13 y 17). De igual manera se debe considerar e incorporar lo establecido en el apartado e) de ese artículo “Existencia de medicamentos u otras alternativas terapéuticas para las mismas afecciones a menor precio o inferior coste de tratamiento”.
CAPÍTULO I
Artículo 2. Definiciones.
1. Enmienda de adición. Art.2.b b) «evaluación de las tecnologías sanitarias»: un proceso multidisciplinar de evaluacióncomparativa de los aspectos médicos, sociales y relativos a las y los pacientes, y lascuestiones económicas y éticas relacionadas con el uso de una tecnología sanitaria demanera sistemática, objetiva, transparente, imparcial y rigurosa;Justificación: La ETS como parte de la toma de decisiones administrativas debenresponder de forma objetiva, mediante medios de prueba, a las cuestiones que venga adar respuesta la evaluación, de forma que, además sea reproducible y otorgue el rigory credibilidad necesario.
2. Enmienda de aclaración. Art.2.f.
f) «desarrollador de tecnologías sanitarias»: persona física o jurídica titular de la autorización de comercialización, responsable de la interlocución con la administración para la incorporación de sutecnología en el sistema nacional de salud.
Justificación: «desarrollador de tecnologías sanitarias»: debe entenderse como el titularde la autorización de comercialización al coexistir de forma habitual variosdesarrolladores en su desarrollo. Aunque el Reglamento 2021/2282 incluye estadefinición, su denominación más adecuada sería el de “titular de la autorización”. En elcaso de que no proceda modificar esta definición acorde al reglamento en cuestión, sesugiere la adición de una enmienda de adición aclaratoria que diga que: f.1) Por desarrollador de la tecnología sanitaria” se entiende al titular de la autorización comercial.
3. Enmienda de adición. Art.2.j.
j) «Evaluación clínica comparada»: proceso multidisciplinar que incluye el análisis comparativo de los datos clínicos disponibles sobre una tecnología sanitaria en comparación otra u otras tecnologías o procedimientos existentes, debiendo incorporar las mejores alternativas existentes para el mismo procedimiento o indicación, de conformidad con un ámbito de evaluación representativo de su posible utilización en el marco del Sistema Nacional de Salud (SNS). Se trata, por tanto, de una evaluación del valor clínico añadido de la nueva tecnología.
Justificación: el valor añadido de la realización la ETS de calidad debe ser el establecer la evidencia de una mejora clínica relevante sobre la tecnología existente para un determinado procedimiento o indicación, para lo que debe compararse con las mejores existentes.
4. Enmienda de adición. Art 2.k.
k) «Evaluación económica comparada»: proceso por el que se analizan los costes de desarrollo e investigación y financiación y la eficiencia de una tecnología paradeterminar los recursos adicionales que exige incorporarla, en relación con losresultados en salud adicionales obtenidos con su adopción en contraste con uncomparador, el cual debe. Otro objetivo es analizar su impacto presupuestario, es decir,los recursos agregados adicionales que exige su adopción. Es un proceso que formaparte de los ámbitos de evaluación no clínicos y debe ser multidisciplinar, objetivo, sistemático, transparente, sin sesgos, y robusto. El comparador debe representar la mejor alternativa clínica existente en el momento de la ETS para la misma afección y la mejor relación coste-eficacia, para lo que debe incluir la existencia de medicamentos u otras alternativas terapéuticas para las mismas afecciones a menor precio o inferior coste de tratamiento.
Justificación: sin conocimiento de los costes de desarrollo, de investigación yfinanciación y el análisis de coste-efectividad es incompleto para adoptar decisiones definanciación e incorporación de tecnología sanitaria en el sistema sanitario, cuyoprocedimiento debe ser comparando estos datos con los de las alternativas existentes.Por otro lado, El artículo 92.1 del RD Legislativo 1/2015 recoge en el apartado e)“Existencia de medicamentos u otras alternativas terapéuticas para las mismasafecciones a menor precio o inferior coste de tratamiento”.
5. Enmienda de adición. Art.2.I
l) «Posicionamiento desde la perspectiva del SNS»: conclusión de la ETS teniendo en cuenta la evaluación clínica comparada, la evaluación económica comparada y, por tanto, su eficiencia, costes de desarrollo, investigación y financiación e impacto presupuestario, así como el resto de los ámbitos de evaluación no clínica en relación con la incorporación de dicha tecnología a la prestación pública de salud. Enmienda de adición (ver justificación de enmienda de adición del apartado k del presente artículo).
6. Enmienda de adición. Nuevo apartado. Art.2.m.
m) Organizaciones de la sociedad civil»: organizaciones sin ánimo de lucro que representan los intereses colectivos de la ciudadanía, de manera independiente del gobierno y de entidades comerciales, y que contribuyen a la promoción de derechos como el derecho a la salud, la participación en la toma de decisiones públicas o la transparencia. Entre ellas, se reconocen de especial relevancia aquellas organizaciones sin ánimo de lucro que agrupan a profesionales de diversos ámbitos” (legislativo, de transparencia y buen gobierno, de información, accesibilidad a los medicamentos) que poseen conocimientos, habilidades, aptitudes y objetivos específicos con un interés en el área del medicamento y de la ETS.
Justificación: Es importante por la aportación de estas organizaciones a la transparencia, a la asignación de recursos y regulación equitativa con una visión diferente y más global que las asociaciones de sanitarios, de pacientes o de consumidores. Ejemplos de organizaciones ciudadanas con interés en el área del medicamento: “Salud por Derecho”, “CIVIO”, “Asociación de Acceso Justo al Medicamento (AAJM)”, “Médicos del Mundo”, etc) La inclusión de la sociedad civil es fundamental para defender derechos humanos en poblaciones que no son representados por otros grupos, pero que se ven afectadas por las ETS, incorporando consideraciones de salud global o impacto en la sostenibilidad del sistema sanitario. Así lo reconoce el artículo 15 del Tratado de Funcionamiento de la Unión Europea, donde se remarca la importancia de la participación de la sociedad civil en la buena gobernanza, y el artículo 11 por el que se enfatiza la necesidad de mantener un diálogo abierto y transparente con las organizaciones de la sociedad civil.
Artículo 4. Consejo de gobernanza.
7. Enmienda. Art.4.i.
i) Una persona miembro de las organizaciones de pacientes, y un miembro de las organizaciones de consumidores y una persona miembro de las organizaciones de la sociedad civil, nombrados por la Secretaría de Estado de Sanidad a propuesta de éstas.
Justificación: Debe especificarse que los participantes de las ETS son miembros de las organizaciones y no personas que éstas puedan designar externamente, lo que desvirtuaría el papel de dichas organizaciones y el valor de sus aportaciones en el proceso de ETS. Del mismo modo, estas organizaciones son las que mejor conocen a sus miembros y las que deben proponer a sus candidatos representantes de las mismas.
8. Enmienda de adición. Nueva. Art.4.k.
k) Las personas nombradas por parte de la administración de los apartados c-h del presente artículo, al margen del desarrollo posterior de un reglamento específico para su selección y nombramiento, deben ser expertas y/o acreditar conocimiento del proceso de ETS, no concurrir en conflictos de intereses, nombradas por periodos de 4 años, cuyo proceso de selección debe ser participativo, público, transparente, independiente, justificado y consensuado, con capacidad de ser propuestas y recusados por las diferentes organizaciones partes de este RD según la concurrencia de conflictos de intereses u otras circunstancias que menoscaben su independencia. Especialmente en cuenta se tendrá la valoración y propuestas por el Consejo Interterritorial de Salud.
Justificación: El Consejo de Gobernanza, como máximo garante y rector del proceso de
ETS debe contar con los mejores expertos y con la mayor independencia posible. Por otro lado, las decisiones adoptadas en el ámbito farmacéutico son en su mayor parte exclusivas del Gobierno Central. Sin embargo, su aplicación depende de las CCAA, siendo el Consejo Interterritorial del SNS donde quedan representadas y exponen sus necesidades en salud y se da respuesta a las mismas.
9. Enmienda de supresión.art. 4.4.c)
Justificación: este objetivo sobrepasa el objetivo y principio rector del Consejo deGobernanza recogido en su el apartado 1 del artículo 4, pudiendo socavar suindependencia y objetividad en el cumplimiento de los mismos, motivo por el cual sejustifica su ausencia como integrante de dicho consejo a las partes interesadas. Por otrolado, ya se dispone y regula el procedimiento de asesorías en el presente RD.
10. Enmienda de adición.art,4.4.d.
d) aprobar, a propuesta de las Oficinas y el Grupo de posicionamiento, las guías metodológicas para el trabajo del «Sistema para la evaluación de la eficiencia de las tecnologías sanitarias», garantizando su alineación con la política farmacéutica y de prestaciones del Ministerio de Sanidad, con los criterios que reglamentariamente configuren la toma de decisiones y siguiendo las normas internacionales de la medicina basada en pruebas y la mejor y más robusta evidencia;
Justificación: una ETS de calidad, que pretenda establecer el valor añadido de la tecnología sanitaria y mejorar la calidad de la innovación, no sólo debe basarse en pruebas, lo que forma parte del proceso en sí más que de la calidad del mismo, sino que debe basarse en la mejor evidencia disponible.
11.Enmienda de adición. Art. 4.5.
5. Para el cumplimiento de sus funciones, el «Consejo de gobernanza» podrá contar de forma justificada y pública con el asesoramiento del Comité Asesor para laPrestación Farmacéutica del Sistema Nacional de Salud. Los miembros de este Comité Asesor que sean consultados deben cumplir con los mismos requisitos sobre conflictos de intereses que el resto de los participantes en los órganos de ETS a los que se refiere este RD.
Artículo 5. Oficinas para la evaluación de la eficiencia de las tecnologías
sanitarias.
12. Enmienda de adición.art.5.1
1. Cada «Oficina para la evaluación de la eficiencia de las tecnologías sanitarias» está formada por todas aquellas estructuras, organismos u organizaciones que participan en la ETS, medicamentos en un caso y resto de tecnologías sanitarias en el otro, con los fines contemplados en el presente real decreto. En concreto se compondrá con una estructura similar a la del Consejo de Gobernanza, incluyendo dos representantes en su apartado j); la propuesta enmendada que incorpora a las organizaciones de la sociedad civil.
Justificación: En aras de la transparencia, objetividad e independencia en la toma de decisiones de este órgano, deben explicitarse y conocerse su composición, así como cumplir con todos los requisitos exigibles a los componentes del resto de órganos del sistema de ETS.
13. Enmienda de adición.art.5.3. párrafo 1.
3. La «Oficina para la evaluación de la eficiencia de los Medicamentos» se configurará como una unidad funcional adscrita al Ministerio de Sanidad. De acuerdo con la disposición adicional tercera de la Ley 10/2013 de 24 de julio, por la que se incorporan al ordenamiento jurídico español las Directivas 2010/84/UE del Parlamento Europeo y del Consejo, de 15 de diciembre de 2010, sobre farmacovigilancia, y 2011/62/UE del Parlamento Europeo y del Consejo, de 8 de junio de 2011, sobre prevención de la entrada de medicamentos falsificados en la cadena de suministro legal, y se modifica la Ley 29/2006, de 26 de julio, de garantías y uso racional de los medicamentos y productos sanitarios y el artículo 31.3 de la Ley 16/2003, de 28 de mayo, de cohesión y calidad del Sistema Nacional de Salud. En lo que se refiere a la evaluación de medicamentos, debe garantizarse la autonomía funcional, la independencia y la ausencia de conflictos de interés de esta unidad funcional con la AEMPS.
Justificación: La AEMPS, como la EMA a nivel europeo, son las responsables de autorizar un nuevo medicamento y se trata por tanto en ambos casos de agencias fundamentalmente reguladoras del medicamento. La cuestión es que si la competencia en la evaluación se adscribe a una Oficina de la AEMPS, que de esta forma asumirá reevaluar el medicamento que ya ha evaluado en el marco regulatorio. Los conflictos de interés de las agencias reguladoras con la industria se derivan de su propia misión, que comporta una relación continua entre ambas en los procesos de autorización y aprobación. Hay una amplia gama de temas regulatorios en que el asesoramiento de la agencia reguladora es esencial.
A la Industria le puede interesar que las conclusiones de una evaluación se conformen con unas indicaciones de tipo genérico similares a la indicación autorizada. Por otro lado, para la Agencia reguladora no resulta cómodo que sus expertos cuestionen lo que ella misma ha aprobado, ni hagan restricciones o cuestionamientos sobre la indicación autorizada por la misma agencia reguladora en la que participan. Recordar que La EMA está financiada por la industria farmacéutica, a su vez, la agencia paga a las autoridades nacionales por la evaluación científica de las solicitudes.
Puigventós F y Alegre E. La cuarta garantía en la evaluación de medicamentos. Revista
de la AAJM 27: 5-15 Feb 2024 https://accesojustomedicamento.org/la-cuarta-garantia-
en-la-evaluacion-de-medicamentos/
14. Enmienda de adición. Art.5.3 párrafo 2º.
La Agencia Española de Medicamentos y Productos Sanitarios representará a España en el Grupo de Coordinación sobre Evaluación de Tecnologías Sanitarias de los Estados miembros, y nombrará a las personas representantes, miembros de las administraciones públicas parte de las Agencias de ETS, en los subgrupos de trabajo que emanan del mismo de acuerdo con el Reglamento (UE) 2021/2282 del Parlamento Europeo y del Consejo, de 15 de diciembre de 2021, para los temas relacionados con medicamentos.
Justificación: Como representantes del Estado, debe tratarse de miembros de la administración pública y, en concreto, de las agencias de ETS.
15. Enmienda de adición. Nuevo.art.5.3. párrafo 3.
La elección de los expertos componentes en la Oficina debe evitar los conflictos de interés derivados de la participación en la oficina de profesionales que al mismo tiempo tengan una función en la evaluación para la autorización del medicamento propio de la agencia reguladora o bien derivadas de su participación en asesorías solicitadas a las agencias reguladoras AEMPS o EMA por parte de la compañía productora de medicamento.
Justificación:Puigventós F y Alegre E. La cuarta garantía en la evaluación demedicamentos. Revista de la AAJM 27: 5-15 Feb 2024https://accesojustomedicamento.org/la-cuarta-garantia-en-la-evaluacion-de-
16. Enmienda de adición. Nuevo.art.5.3. párrafo 4.
4. La composición de La «Oficina para la evaluación de la eficiencia de los Medicamentos» debe tener una estructura similar del Consejo de Gobernanza, cuyas personas nombradas por parte de la administración de los apartados c-h y j, al margen del desarrollo posterior de un reglamento específico para su selección y nombramiento, deben ser expertas y/o acreditar conocimiento del proceso de ETS, no concurrir en conflictos de intereses, nombradas por periodos de 4 años, cuyo proceso de selección debe ser participativo, público, transparente, independiente, justificado y consensuado, con capacidad de ser propuestas y recusados por las diferentes organizaciones partes de este RD según la concurrencia de conflictos de intereses u otras circunstancias que menoscaben su independencia. Especialmente en cuenta se tendrá la valoración y propuestas por el Consejo Interterritorial de Salud. En el apartado j) de dicha composición serán dos las personas nombradas.
Justificación: la evaluación económica debe respetar especialmente y de forma escrupulosa su independencia e imparcial, con la ausencia total de conflictos de intereses, por lo que su la definición de su estructura y la publicidad de todo lo relativo a los miembros que la componente y actividades es imprescindible. Se hace una propuesta de composición de este órgano, la cual no está definida en el RD. Por otro lado, las decisiones adoptadas en el ámbito farmacéutico son en su mayor parte exclusivas del Gobierno Central. Sin embargo, su aplicación depende de las CCAA, siendo el Consejo Interterritorial del SNS donde quedan representadas y exponen sus necesidades en salud y se da respuesta a las mismas.
17. Enmienda de modificación. Art.5.5.a. a) elaborar la evaluación clínica comparada y evaluación económica comparada* deacuerdo con lo contemplado en los artículos 8 a 12 13 que permita una adecuadavaloración de las tecnologías sanitarias la toma de decisiones;
Justificación: Debe haber una errata en el artículo 5.5 a) que dice “elaborar la evaluaciónclínica comparada y evaluación económica de acuerdo a lo contemplado en los artículos8 a 12 …”. Creemos que se refiere a los artículos 8 a 13. Ver justificación de enmienda4*.
18. Enmienda de supresión. Art.5.5.b.
Justificación: se entiende que la asesoría debe ser eminentemente clínica y/orelacionada con la investigación e innovación, por lo que sobrepasa las competencias yobjetivos económicos que pretende y compete a este órgano.
19. Enmienda de adición. Art.5.5.f.
f) proponer las guías metodológicas para el trabajo de evaluación, garantizando su alineación con la política farmacéutica y de prestaciones del Ministerio de Sanidad, con los criterios que reglamentariamente configuren la toma de decisiones y siguiendo las normas internacionales de la medicina basada en pruebas y la mejor y más robusta evidencia, y que aseguren la coherencia con la ETS a nivel europeo según el Reglamento (UE) 2021/2282 del Parlamento Europeo y del Consejo, de 15 de diciembre de 2021, sobre evaluación de las tecnologías sanitarias dar cuenta de la metodología en el trabajo de evaluación y de los criterios de categorización del valor clínico añadido.
Justificación: una ETS de calidad, que pretenda establecer el valor añadido de la tecnología sanitaria y mejorar la calidad de la innovación, no sólo debe basarse en pruebas, lo que forma parte del proceso en sí más que de la calidad de este, sino que debe basarse en la mejor evidencia disponible.
Artículo 6. Grupo de posicionamiento de las tecnologías sanitarias.
20. Enmienda de modificación.art.6.3.i.
i) Una persona miembro de las organizaciones de pacientes, y miembro de las organizaciones de consumidores y una persona miembro de las organizaciones de la sociedad civil, nombrados por la Secretaría de Estado de Sanidad a propuesta de éstas. Justificación: Debe especificarse que los participantes de las ETS son miembros de las organizaciones y no personas que éstas puedan designar externamente, lo que desvirtuaría el papel de dichas organizaciones y el valor de sus aportaciones en el proceso de ETS. Del mismo modo, estas organizaciones son las que mejor conocen a sus miembros y las que deben proponer a sus candidatos representantes de las mismas.
21. Enmienda de adición.art.6.7.
7. De forma excepcional y debidamente justificado, para aspectos concretos, el «Grupo de posicionamiento» de las tecnologías sanitarias podrá contar con la participación ad hoc de personas expertas, profesionales y/o pacientes, que podrán participar en las discusiones con voz, pero sin voto.
Justificación: garantizar la mayor objetividad e independencia en la toma de decisiones basada en los resultados de las evaluaciones realizadas.
22. Enmienda de adición. Art.6.8.
8. De forma excepcional, pública y debidamente justificada, para aspectos concretos, los desarrolladores de tecnologías podrán tener audiencia con el grupo deposicionamiento de las tecnologías sanitarias para solventar aquellas cuestiones que nose encuentren recogidas en el informe de evaluación y que puedan ser relevantes para la toma de decisiones facilitar la discusión con todos los interlocutores.
Justificación: se debe garantizar la mayor imparcialidad, objetividad, independencia y transparencia del proceso.
23. Enmienda de modificación. Art. 6.9.
9. El «Grupo de Posicionamiento» actuará, en principio, por consenso. Cuando no pueda alcanzarse un consenso, la adopción de una decisión requerirá el apoyo de los miembros que representen una mayoría simple de los miembros. Los resultados de las votaciones se harán constar en las actas de las reuniones del «Grupo de Posicionamiento». Cuando se celebre una votación, se harán constar todas las opiniones divergentes en el acta de la reunión en la que se haya celebrado la votación. Justificación: garantías de transparencia y buen gobierno.
24. Enmienda adición. Art.6.10.d.
d) garantizar que sean coherentes con la regulación pertinente de criterios de precio y financiación o de inclusión en la cartera de servicios del Sistema Nacional de Salud y el objetivo de preservar y promover la sostenibilidad del sistema nacional de salud; Justificación: una de las principales justificaciones del RD es el desarrollo de la evaluación de los criterios no clínicos, entre los que se encuentran los económicos, con el objetivo de favorecer la sostenibilidad del sistema nacional de salud.
25. Enmienda de adición.art.6.10.f.
f) adoptar y presentar cada año al «Consejo de gobernanza» un informe anual público, en el que se proporcionará información sobre el trabajo realizado el año natural anteriora su adopción.
Justificación. Garantizar la transparencia y el buen gobierno.
26. Enmienda de adición. Nueva. Art.6.11.
11. Las personas nombradas por parte de la administración del artículo 6 en sus apartados 3.c-g y 4.c-h, al margen del desarrollo posterior de un reglamento específico para su selección y nombramiento, deben ser expertas y/o acreditar conocimiento del proceso de ETS, no concurrir en conflictos de intereses, nombradas por periodos de 4 años, cuyo proceso de selección debe ser participativo, público, transparente, independiente, justificado y consensuado, con capacidad de ser propuestas y recusados por las diferentes organizaciones partes de este RD según la concurrencia de conflictos de intereses u otras circunstancias que menoscaben su independencia. Especialmente en cuenta se tendrá la valoración y propuestas por el Consejo Interterritorial de Salud.
Justificación: El Grupo de Posicionamiento, último decisor en el proceso de evaluación respecto al valor añadido y comparado de la tecnología sanitaria, debe contar con los mejores expertos y con la mayor independencia posible. Por otro lado, las decisiones adoptadas en el ámbito farmacéutico son en su mayor parte exclusivas del Gobierno Central. Sin embargo, su aplicación depende de las CCAA, siendo el Consejo Interterritorial del SNS donde quedan representadas y exponen sus necesidades en salud y se da respuesta a las mismas.
CAPÍTULO II
Artículo 7. Tecnologías objeto de la evaluación de tecnologías sanitarias.
27. Enmienda de adición. Art. 7.1.b.
b) los nuevos medicamentos autorizados por otros procedimientos que se pretendan comercializar en España y en los que se considere necesario realizar un posicionamiento terapéutico en atención a los criterios que serán desarrollados y publicados, de manera consensuada, por el «Consejo de gobernanza» y que tendrán en cuenta la posición y necesidades del Consejo Interterritorial del Sistema Nacional de Salud.
Justificación: Las decisiones adoptadas en el ámbito farmacéutico son en su mayor parte exclusivas del Gobierno Central. Sin embargo, su aplicación depende de las CCAA, siendo el Consejo Interterritorial del SNS donde quedan representadas y exponen sus necesidades en salud y se da respuesta a las mismas.
28. Enmienda de adición. Art.7.1.f.
f) cualquier otro producto sanitario, procedimiento médico o quirúrgico, tecnología digital, guías de práctica clínica y protocolos de actuación o medidas o modelos organizativos para la prevención, el diagnóstico o el tratamiento de enfermedades utilizados en la asistencia sanitaria que sea acordado por el «Consejo de gobernanza»
Justificación: en aras de un uso racional y eficiente de las tecnologías sanitarias que redunde en la mejor práctica sanitaria posible y en la contribución a la sostenibilidad del SNS, a su vez, éste debe dotar de instrumentos necesarios para ello a profesionales y organismos públicos prestadores de los servicios sanitarios, para lo que las guías de práctica clínica constituyen una herramienta fundamental, pero que a día de hoy se elaboran fuera del ámbito donde se deben aplicar y cuyos intereses no siempre se
encuentran alineados.
29. Enmienda de adición. Art. 7 1.g
g) cualquier tecnología sobre la que se pretenda evaluar los resultados en salud.
Justificación: los resultados en salud son la mejor evidencia y medio de prueba del valor clínico añadido de la tecnología sanitaria.
30. Enmienda de adición. Nueva. Art. 7 1. h
h) aquellas que se evalúen con el objetivo de un proceso de desinversión.
Justificación: la información de los procesos de desinversión es uno de los objetivos dela ETS.
31. Enmiendas de adición. Nuevas: Art. 7. 2. g, h, i, k.
g) terapias avanzadas, especialmente las derivadas de la aplicación de tecnología basadas en la genética.
h) se prevea un impacto importante impacto presupuestario.
i) una calidad innovativa relevante.
j) tecnología objeto de desinversión.
k) evaluación según resultados en salud post-autorización comercial.
Artículo 8. Contenido de las evaluaciones.
32. Enmienda de adición. Art 8.4.
4. La evaluación económica incluirá, además de la evaluación de la eficiencia, los costes de desarrollo, investigación y financiación de la tecnología sanitaria, una aproximación de su impacto presupuestario y aportará el análisis necesario según el caso siguiendo la metodología aprobada por el Consejo de Gobernanza que permita establecer al menos la eficiencia de la nueva tecnología.
Justificación: sin conocimiento de los costes, el análisis de coste-eficiencia es incompleto para adoptar decisiones de financiación e incorporación de tecnología sanitaria en el sistema sanitario, cuyo procedimiento debe ser comparando estos datos con los de las alternativas existentes. Este requerimiento es coherente con el artículo 92. del RD Legislativo 1/2015.
Artículo 9. Aspectos generales de la evaluación clínica.
33. Enmienda de modificación. Art. 9.1.
1. Para determinar la efectividad comparada se utilizarán en cuenta las mejores y más eficientes alternativas asistenciales existentes u y aquellas otras alternativasfacilitadas actualmente dentro del Sistema Nacional de Salud para la afección a la que se dirige.
Justificación: el valor añadido de la realización la ETS de calidad debe ser el establecer la evidencia de una mejora clínica relevante sobre la tecnología existente para un determinado procedimiento o indicación, para lo que debe compararse con las mejores
existentes.
34. Enmienda de adición. Nueva. Art. 9.2.e)
e) el proceso clínico, problema de salud o afección al que se dirige.
Justificación: un aspecto fundamental del ámbito donde se pretende evaluar la utilidad y valor añadido de una tecnología es el proceso clínico, problema de salud o afección para el que se pretende indicar la tecnología en cuestión.
35. Enmienda de modificación. Art. 9.4.
4. Las evaluaciones se basarán un expediente que contenga informaciones, datos,análisis y otros elementos de prueba completos y actualizados, presentado por eldesarrollador de tecnologías sanitarias u otros que el órgano evaluador considere relevante para el proceso, ya sean requeridos al desarrollador o disponibles por otros medios obtenidos por otros medios, para evaluar los parámetros incluidos en el ámbito de evaluación previamente definido.
Justificación: los datos y medios de prueba necesarios para una ETS de calidad y completa deben ser todos los existentes y más oportunos para ello.
36. Enmienda de adición. Nueva. Art. 9.4.1.
4.1. La oposición u omisión de datos por parte del desarrollador puede ser causa de paralización de la evaluación o de su anulación si se hubiera concluido.
Justificación: los datos y medios de prueba necesarios para una ETS de calidad y completa deben ser todos los existentes, fidedignos y los más oportunos para ello.
37. Enmienda de adición. Art. 9.7.
7. Los informes de evaluación clínica incluirán una descripción clara de sus efectos beneficiosos, serán públicos y contendrán un resumen de todas las opiniones emitidas, nivel de evidencia y las conclusiones sobre el valor clínico añadido global de la tecnología sanitaria evaluada, pero no recomendaciones para la posterior toma de decisiones.
Justificación: el grado de evidencia de los resultados de la evaluación son importantes en la toma de decisiones sobre la incorporación y financiación de una determinada tecnología sanitaria en el SNS. El proceso de ETS debe entenderse como un proceso vivo y permanente donde es importante conocer los diferentes puntos de vistas que en otro momento pudieran alcanzar un mayor nivel de evidencia con el surgimiento de nuevas informaciones. Una descripción clara de los efectos beneficiosos es imprescindible puesto que la mayoría de los fármacos nuevos solo ofrecen un pequeño beneficio adicional con respecto a otros ya disponibles. Y aunque se difunde el análisis minucioso de los datos de los ensayos clínicos, los documentos más importantes son inescrutables para casi cualquiera.
38. Enmienda de adición. Art. 9.7.1.
7.1. En las actas de las sesiones de ETS se recogerán todas las opiniones y fundamentaciones sobre la misma.
Justificación: El proceso de ETS debe entenderse como un proceso vivo y permanente donde es importante conocer los diferentes puntos de vistas que en otro momento pudieran alcanzar un mayor nivel de evidencia con el surgimiento de nuevas informaciones.
Artículo 10. Aspectos específicos de la evaluación clínica de los medicamentos.
39. Enmienda de adición. Nuevo. Art.10.1.j)
j) los datos en vida real pertinentes y existentes según proceso de ETS como la desinversión.
Justificación: Justificación: los resultados en salud son la mejor evidencia y medio de prueba del valor clínico añadido de la tecnología sanitaria.
Artículo 13. Evaluación de los aspectos no clínicos.
40.Enmienda de adición. Art. 13.2.
2. La evaluación económica ofrecerá información útil para la toma de decisiones en el marco que se establece reglamentariamente mediante una evaluación robusta que considere el valor de la tecnología sanitaria desde la perspectiva de la efectividad relativa, el valor social de la tecnología sanitaria, el impacto sobre la calidad de vida relacionada con la salud, sus costes de desarrollo, investigación y financiación, su análisis de coste-efectividad, el impacto presupuestario sobre el SNS y la comparación entre el coste real y el precio propuesto o el precio aprobado en otros países de la UE . Esta información identificará la eficiencia de la nueva tecnología en comparación con las alternativas disponibles, así como el análisis de su impacto presupuestario.
Justificación: el conocimiento de los costes de desarrollo, de investigación y financiación, el análisis de coste-efectividad son imprescindibles en una evaluación económica rigurosa, así como el potencial impacto presupuestario en el SNS, cuya sostenibilidad es uno de los objetivos centrales del RD. Una evaluación económica incompleta puede ser perjudicial para adoptar decisiones de financiación e incorporación de tecnología sanitaria en el sistema sanitario. Los acuerdos de confidencialidad de las negociaciones de los diferentes Estados Miembros de la UE con las compañías farmacéuticas determinan diferencias importantes entre el precio público y el pagado por las administraciones públicas, a su vez, diferente en cada Estado, sin que se relacione con la riqueza del país, según la literatura reciente, ni con los costes de desarrollo de los mismos, sino con la disposición a pagar, entre otras circunstancias.
Artículo 14. Plazos de las evaluaciones.
41. Enmienda de modificación. Art. 14.5.
5. Las «Oficinas para la evaluación de la eficiencia de las tecnologías sanitarias» velarán para que los personas pacientes, organizaciones de la sociedad civil en el ámbito del derecho a la portección de la salud, las personas expertas clínicas y otras expertas pertinentes puedan involucrarse a título individual o colectivamente en el proceso de evaluación dándoles la oportunidad de hacer aportaciones a los proyectos de informe. Dichas aportaciones se presentarán dentro del marco y en los plazos establecidos que se determinen en el Documentos de instrucciones para la ETS en España que se recoge en el artículo 22.
Justificación: el proceso debe garantizar la partición amplia de todos aquellas partes que puedan aportar y enriquecer el proceso de evaluación en aras de mejorar su calidad con el mayor conocimiento posible y disponible.
42. Enmienda de modificación. Art. 14.6
6. Los informes de evaluación, tras de su finalización y previo a su publicación, podrán ser consultados con el fin de señalar cualquier inexactitud meramente técnica o fáctica dentro de los plazos establecidos en el Documentos de instrucciones para la ETS en España que se recoge en el artículo 22. Seña lará asimismo cualquier información que considere confidencial y justificará su carácter comercialmente sensible. No hará observaciones sobre los resultados del proyecto de evaluación.
Justificación: los defectos técnicos y aspectos confidenciales a señalar no deben modificar ninguna decisión sobre la evaluación, por lo que ésta debe estar concluida.
Artículo 15. Sobre los efectos de las evaluaciones.
43. Enmienda de adición. Art. 15.4.
4. El resultado de una evaluación puede estar condicionado a la resolución de las incertidumbres que hayan sido identificadas en el transcurso de la misma, pudiendo proponer el modelo o modelos de cómo generar la evidencia que permitan solventar estas incertidumbres. Para ello, se fomentará el dialogo temprano con los desarrolladores en el marco de las consultas de asesoría a que se hace referencia en el artículo 19. El resultado de tales consultas debe ser incluido en el informe de evaluación.
Justificación: deben conocerse todas las decisiones tenidas en cuenta en la ETS en aras de la independencia, objetividad, rigor y transparencia que avalen el procese de ETS y las decisiones de la cual se deriven.
Artículo 16. Publicación de las evaluaciones.
44. Enmienda de modificación. Art. 16.4.1.
1. Los informes de evaluación de las «Oficinas para la evaluación de la eficiencia de las tecnologías sanitarias» serán públicos salvo aquellos aspectos confidenciales debidamente justificados y que, no obstante, formarán parte del documento de evaluación.
Justificación: aplicación del artículo 97.3 del Real Decreto Legislativo 1/2015 en aras de una mayor transparencia.
Artículo 18. Uso de datos y datos en vida real.
45.Enmienda de adición art. 18.1
1. Se posibilitará el uso de datos reales en salud para aprovechar el potencial que ofrece el intercambio, uso y reutilización de datos sanitarios en beneficio de las personas pacientes, las investigadoras, las innovadoras y las reguladoras, garantizando y dirigidos a preservar la sostenibilidad del sistema nacional de salud mediante un sistema de criterios de retorno de la inversión pública en precios asequibles y promoviendo la investigación pública a través de una plataforma pública nacional para la I+D en salud, dentro de un sistema coherente, fiable, eficiente y garantista de los derechos fundamentales de los pacientes, siguiendo la normativa pertinente según el Reglamento europeo del Espacio de datos en salud y la correspondiente normativa de desarrollo nacional. No obstante, el uso de estos datos de salud y su que forman parte del SNS deben redundar en garantizar su sostenibilidad, sin suponer un sobrecoste para las arcas públicas y/o barreras en el acceso de las tecnologías derivadas de su manejo, por lo que debe garantizarse el retorno de la inversión pública.
Justificación: La oportunidad de “explotación” y aprovechamiento del conocimiento que ofrecen las nuevas tecnologías en el manejo de los datos sobre la salud suponen una importante oportunidad de impulso a la I+D, como lo recoge y justifica el reglamento europeo sobre el espacio europeo de datos sanitarios; pero también y, sobre todo, debe apoyar y garantizar la sostenibilidad del SNS, titular, proveedor y depositario de los mismos. Estos datos que forman parte del SNS y que se han generado desde un sistema solidario de financiación pública fiscalmente progresiva, deben redundar en garantizar su sostenibilidad y no en el enriquecimiento privado y, menos aún, suponer un sobrecoste para las arcas públicas y/o barreras en el acceso de las tecnologías derivadas de su manejo, por lo que debe garantizarse el retorno de la inversión pública y prevalecer el interés general superior que representa un SNS sostenible y robusto para hacer efectivo el Derecho Fundamental a la Salud y, en concreto, a las tecnologías sanitarias, evitando cualquier forma de descapitalización del mismo o ser origen de desigualdades en su acceso.
46.Enmienda de sustitución Art. 18.3
3. El Ministerio de Sanidad mantendrá garantizará sistemas de información que permitan evaluar y monitorizar los resultados del «Sistema para la evaluación de la eficiencia de las tecnologías sanitarias» de forma garante y respetuosa con los derechos fundamentales de los pacientes.
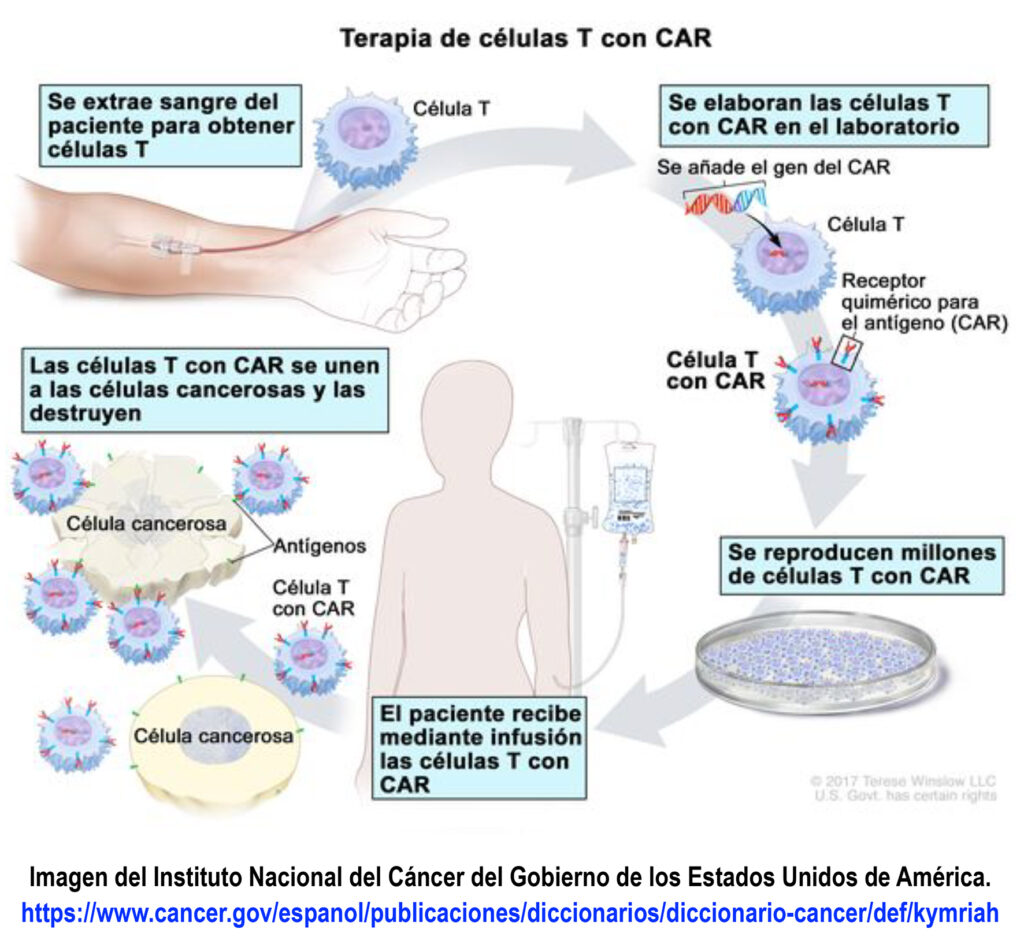
Justificación: el proceso de ETS de calidad debe ser dinámico y actualizado, para lo que precisa de la mayor evidencia posible, para lo cual deben ponerse los medios necesarios al alcance del SNS, al mismo tiempo que la información sobre los datos reales en salud suponen una gran oportunidad para incentivar la I+D pública. La irrupción de nuevas tecnologías que permiten el manejo de grandes volúmenes de datos o la inteligencia artificial, junto con los avances en disciplinas como la genética y la biotecnología, en general, abren una oportunidad para que un sistema público de investigación pueda dar respuesta accesible y asequible a nuevas terapias; enmarcadas muchas de ellas en lo que se ha venido a denominar “medicina de precisión”. Un importante ejemplo de ello son las terapias CAR-T, en las que España, gracias a la exención hospitalaria, entre otras medidas, se sitúa a la vanguardia. Por otro lado, la incertidumbre que genera la rápida evolución de las tecnologías digitales y su capacidad de control debe ser vigilante y garantista con los Derechos Fundamentales de la ciudadanía.
CAPÍTULO III
Artículo 19. Consultas de asesoría.
47. Enmienda de modificación. Art.19.7.
7. Las Oficinas incluirán información resumida anonimizada, sin anonimizar, y agregada desagregada, excluyendo la información que se considere confidencial y no confidencial sobre las consultas de asesoría en sus informes anuales y en suspáginas web. En todo caso, las consultas de asesoría se regirán por los principios departicipación, transparencia y conflicto de interés que se recogen en los artículos 24, 25y 26.
Justificación: Garantizar la independencia y transparencia del sistema de ETS.
48. Enmienda de modificación. Art.19.8.
8. Las consultas de asesoría estarán sujetas a una tasa o precio público, según corresponda, que se fijarán de acuerdo a la Ley.
Justificación: No se deben fijar tarifas, para garantizar la independencia del sistema de ETS. Su financiación, como la de todo lo relativo al proceso de ETS debe llevarse a cabo con el desarrollo adecuado, suficiente y actualizado de la disposición adicional sexta relativa al sistema de tasas por ventas en el SNS por parte de los desarrolladores de tecnología sanitaria.
CAPÍTULO IV
Artículo 23. Obligaciones de los desarrolladores de tecnologías sanitarias y
consecuencias del incumplimiento.
49. Enmienda de adición. Art. 23.7.
7. El desarrollador de una tecnología en evaluación está obligado a aportar los costes fidedignos de producción, investigación y desarrollo, así como de las fuentes de financiación de estos costes, públicas o privadas e incentivos fiscales, de forma desagregada, completa y fidedigna. Del mismo modo, presentará el coste de adquisición de la tecnología sanitaria si no es su desarrollador original. La no aportación por parte del desarrollador de esta información o parte de ella suspenderá el proceso de evaluación de esa tecnología hasta que la aporte y/o subsane.
Justificación: según los datos de la OCDE sobre I+D pública y privada para 2018, la financiación pública es ya superior al 40%, al menos, del mismo modo que el Informe realizado por la Consultora Copenhague encargado por la Comisión Europea para evaluación del funcionamiento del sistema de incentivos a la I+D farmacéutica, estima que el de la UE es el más generoso. Todo ello no sólo debería abaratar los costes de desarrollo de las tecnologías sanitarias, sino que deberían reflejarse en los precios de las mismas de forma que el retorno de la inversión pública sea en términos clínicos, sociales y económicos. Por otro lado, cada vez es más frecuente que las grandes compañías farmacéuticas compren pequeñas empresas con los productos en desarrollo, tras las primeras fases de la investigación, y que sólo completen los estudios en las fases avanzadas de los ensayos clínicos. A nivel de las empresas de biotecnología, este proceso es un fenómeno especialmente creciente, y que se ha venido a denominar “empresas derivadas”, las cuales han surgido en la mayoría de los casos de las universidades públicas y posteriormente son adquiridas por las grandes compañías en un proceso de transferencia de tecnología.
50. Enmienda de adición. Art. 23.8.a).
a) los elementos de prueba presentados deberán ser completos en relación con todoslos estudios y datos disponibles que puedan fundamentar la evaluación;
Justificación: en la ETS de calidad se debe disponer y realizar con la mayor y mejor información disponible.
Artículo 24. Garantía de calidad.
51. Enmienda de adición. Art.24.1.
1.El «Sistema para la evaluación de la eficiencia de las tecnologías sanitarias» garantizará que el trabajo conjunto llevado a cabo sea de la máxima calidad, siga las normas internacionales de la medicina basada en pruebas y la mejor y más robusta evidencia y se ejecute en tiempo oportuno.
Justificación: una ETS de calidad, que pretenda establecer el valor añadido de la tecnología sanitaria y mejorar la calidad de la innovación, no sólo debe basarse en pruebas, lo que forma parte del proceso en sí más que de la calidad del mismo, sino que debe basarse en la mejor evidencia disponible en el momento de la evaluación.
Artículo 25. Garantías de participación de las personas pacientes, consumidoras
y profesionales.
52. Enmienda de modificación. Art. 25. 1.
1. Se garantizará la participación sistemática, cuando proceda con voz y voto, de las personas miembros de las organizaciones de pacientes, organizaciones de consumidores, organizaciones de la sociedad civil y profesionales sanitarias y nosanitarias, si procede, en las actividades recogidas en este real decreto para incorporarsu perspectiva en todas ellas. Sus aportaciones serán recogidas en el informe de evaluación y serán públicas, debiéndose tener en cuenta de forma pertinente.
Justificación: la participación de la sociedad en sus diferentes manifestaciones ajenas a la administración, al poder legislativo y ejecutivo, vienen a garantizar la necesaria democratización de las decisiones adoptadas.
53. Enmienda de modificación. Art. 25. 2.
2. El Ministerio de Sanidad publicará la metodología aprobada por el Consejo de Gobernanza a propuesta de las Oficinas y el Grupo de Posicionamiento para proponer, seleccionar, incorporar y hacer efectiva la participación de organizaciones de pacientes, organizaciones de consumidores, organizaciones de la sociedad civil y organizaciones profesionales en las actividades que les corresponden.
Justificación: Es importante por la aportación de estas organizaciones a la transparencia, a la asignación de recursos y regulación equitativa con una visión diferente y más global que las asociaciones de sanitarios, de pacientes o de consumidores. Ejemplos de organizaciones ciudadanas con interés en el área del medicamento: “Salud por Derecho”, “CIVIO”, “Asociación de Acceso Justo al Medicamento (AAJM)”, “Médicos del Mundo”, etc). La inclusión de la sociedad civil es fundamental para defender derechos humanos en poblaciones que no son representados por otros grupos, pero que se ven afectadas por las ETS, incorporando consideraciones de salud global o impacto en la sostenibilidad del sistema sanitario. Así lo reconoce el artículo 15 del Tratado de Funcionamiento de la Unión Europea, donde se remarca la importancia de la participación de la sociedad civil en la buena gobernanza, y el artículo 11 por el que se enfatiza la necesidad de mantener un diálogo abierto y transparente con las organizaciones de la sociedad civil.
54. Enmienda de adición. Art. 25.3
3. Las organizaciones de pacientes, organizaciones de consumidores, organizaciones de la sociedad civil y organizaciones profesionales deberán cumplir los siguientescriterios:
Justificación: Es importante por la aportación de estas organizaciones a la transparencia, a la asignación de recursos y regulación equitativa con una visión diferente y más global que las asociaciones de sanitarios, de pacientes o de consumidores. Ejemplos de organizaciones ciudadanas con interés en el área del medicamento: “Salud por Derecho”, “CIVIO”, “Asociación de Acceso Justo al Medicamento (AAJM)”, “Médicos del Mundo”, etc). La inclusión de la sociedad civil es fundamental para defender derechos humanos en poblaciones que no son representados por otros grupos, pero que se ven afectadas por las ETS, incorporando consideraciones de salud global o impacto en la sostenibilidad del sistema sanitario. Así lo reconoce el artículo 15 del Tratado de Funcionamiento de la Unión Europea, donde se remarca la importancia de la participación de la sociedad civil en la buena gobernanza, y el artículo 11 por el que se enfatiza la necesidad de mantener un diálogo abierto y transparente con las organizaciones de la sociedad civil.
55. Enmienda de adición. Art.25.3.d)
d) representar los intereses de las personas pacientes, consumidoras o profesionales o que sirvan al interés general a través de la defensa del derecho a la salud,
Justificación: Es importante por la aportación de estas organizaciones a la transparencia, a la asignación de recursos y regulación equitativa con una visión diferente y más global que las asociaciones de sanitarios, de pacientes o de consumidores. Ejemplos de organizaciones ciudadanas con interés en el área del medicamento: “Salud por Derecho”, “CIVIO”, “Asociación de Acceso Justo al Medicamento (AAJM)”, “Médicos del Mundo”, etc). La inclusión de la sociedad civil es fundamental para defender derechos humanos en poblaciones que no son representados por otros grupos, pero que se ven afectadas por las ETS, incorporando consideraciones de salud global o impacto en la sostenibilidad del sistema sanitario. Así lo reconoce el artículo 15 del Tratado de Funcionamiento de la Unión Europea, donde se remarca la importancia de la participación de la sociedad civil en la buena gobernanza, y el artículo 11 por el que se enfatiza la necesidad de mantener un diálogo abierto y transparente con las organizaciones de la sociedad civil.
56. Enmienda de adición. Nuevo. Art.25.3.h)
h) no presentar conflictos de interés ni financiación por parte de entidades relacionadas o con el área de trabajo o tecnología objeto de evaluación. No podrán formar parte del proceso de evaluación aquella organización que hayan recibido cualquier tipo de pago por parte del desarrollador de la ETS en los 5 años anteriores.
Justificación: evitar los conflictos de intereses en aras de garantizar la independencia e imparcialidad de las evaluaciones.
57. Enmienda de adición. Art.25.3. repetido que debería corresponder a un 4 y así sucesivamente los apartados el 5 y 6.
3. 4. Para concretar la participación de las personas pacientes, consumidoras, miembros de las organizaciones de la sociedad civil o profesionales sanitarias o, cuando proceda, no sanitarias, de manera individual en las evaluaciones o grupos de trabajo, se solicitará a las organizaciones relacionadas con el tema a abordar en cada momento, que designen a un miembro de su organización representante experto para formar parte del grupo de trabajo. Esta persona deberá cumplir los siguientes requisitos:
Justificación: La inclusión de la sociedad civil es fundamental para defender derechos humanos en poblaciones que no son representados por otros grupos, pero que se ven afectadas por las ETS, incorporando consideraciones de salud global o impacto en la sostenibilidad del sistema sanitario. Así lo reconoce el artículo 15 del Tratado de Funcionamiento de la Unión Europea, donde se remarca la importancia de la participación de la sociedad civil en la buena gobernanza, y el artículo 11 por el que se enfatiza la necesidad de mantener un diálogo abierto y transparente con las organizaciones de la sociedad civil.Debe especificarse que los participantes de las ETS son miembros de las organizaciones y no personas que éstas puedan designar externamente, lo que desvirtuaría el papel de dichas organizaciones y el valor de sus aportaciones en el proceso de ETS. Del mismo modo, estas organizaciones son las que mejor conocen a sus miembros y las que deben proponer a sus candidatos representantes de las mismas.
58. Enmienda de adición. Nuevo. Art.25.3.c, que debería ser 4.c.
c) no presentar conflictos de interés ni haber recibido cualquier tipo de pago por parte del desarrollador de la ETS en los 5 años anteriores.
Justificación: evitar los conflictos de intereses en aras de garantizar la independencia e imparcialidad de las evaluaciones.
Artículo 26. Garantías de transparencia y conflicto de intereses.
59. Enmienda de adición. Art.26.3.
3. Los participantes en el «Sistema para la evaluación de la eficiencia de las tecnologías sanitarias» no tendrán interés económico o de otro tipo en la industria de los desarrolladores de tecnologías sanitarias que pueda afectar a su independencia o imparcialidad. Se considerarán conflicto de interés la participación en actividades de asesoría científica, estratégica, percepción económica o de cualquier otro tipo realizada a la industria de los desarrolladores de forma directa o indirecta a través de actividades de consultoría. Las directrices de participación de terceros en la ETS detallarán los posibles conflictos de interés incompatibles con la evaluación.
Justificación: evitar los conflictos de intereses en aras de garantizar la independencia e imparcialidad de las evaluaciones.
60. Enmienda de adición. Nuevo. Art.26.3.1
3.1) las organizaciones o cualquier miembro o persona participante en el proceso de ETS no deben presentar conflictos de interés ni financiación por parte de entidades relacionadas o con el área de trabajo o tecnología objeto de evaluación a lo largo de los 5 años anteriores.
Justificación: evitar los conflictos de intereses en aras de garantizar la independencia e imparcialidad de las evaluaciones.
61. Enmienda de adición. Nuevo. Art.26. 9.
9.El Ministerio de Sanidad elaborará un procedimiento garantista por el que los miembros y organizaciones participantes en el proceso de ETS puedan ser recusados, garantizando el anonimato de las personas que revelen los posibles conflictos de interés a investigar.
Justificación: evitar los conflictos de intereses en aras de garantizar la independencia e imparcialidad de las evaluaciones.
Artículo 27. Financiación del «Sistema para la evaluación de la eficiencia de las
tecnologías sanitarias».
62. Enmienda de adición. Art.27.2.
2. El «Sistema para la evaluación de la eficiencia de las tecnologías sanitarias» se podrá financiar a través del sistema de aportaciones por volumen de ventas al Sistema Nacional de Salud recogido en la disposición adicional sexta del Real Decreto Legislativo 1/2015, de 24 de julio, por el que se aprueba el texto refundido de la Ley de garantías y uso racional de los medicamentos y productos sanitarios., a través de los presupuestos generales del Estado, y/o mediante un sistema basado en tasas o precios públicos el cual debe ser revisado para conducirlo hacia una adecuada proporcionalidad y coherencia con las ganancias y margen de beneficios medio de los desarrolladores del resto de sectores industriales.
Justificación: garantizar la independencia y ausencia de conflicto de intereses de la ETS, así como para apoyar la suficiencia de financiación y sostenibilidad del SNS, debiendo dirigirse hacia la proporcionalidad y coherencia con el resto de sectores industriales.
Enmienda de adición. Disposición adicional
63. Enmienda de adición. Disposición transitoria primera.
Disposición transitoria primera. Régimen transitorio relativo a la selección de personas pacientes, consumidoras y profesionales para participar en los grupos de trabajo.
En tanto se desarrollan y publican las directrices para proponer, seleccionar, incorporar y hacer efectiva la participación de organizaciones de pacientes, organizaciones de consumidores, organizaciones de la sociedad civil y organizaciones profesionales en la ETS, las personas representantes de cada uno de estos grupos serán nombradas porlos organismos responsables del funcionamiento de los grupos de trabajo en los que van a participar.
Justificación: Es importante por la aportación de estas organizaciones a la transparencia, a la asignación de recursos y regulación equitativa con una visión diferente y más global que las asociaciones de sanitarios, de pacientes o de consumidores. Ejemplos de organizaciones ciudadanas con interés en el área del medicamento: “Salud por Derecho”, “CIVIO”, “Asociación de Acceso Justo al Medicamento (AAJM)”, “Médicos del Mundo”, etc).La inclusión de la sociedad civil es fundamental para defender derechos humanos en poblaciones que no son representados por otros grupos, pero que se ven afectadas por las ETS, incorporando consideraciones de salud global o impacto en la sostenibilidad del sistema sanitario. Así lo reconoce el artículo 15 del Tratado de Funcionamiento de la Unión Europea, donde se remarca la importancia de la participación de la sociedad civil en la buena gobernanza, y el artículo 11 por el que se enfatiza la necesidad de mantener un diálogo abierto y transparente con las organizaciones de la sociedad civil.
64. Enmienda de adición. Nueva. Disposición adicional primera.
El Ministerio de Sanidad pondrá en marcha, a más tardar, en el plazo de un año tras la entrada en vigor del presente real decreto, un sistema público de transparencia donde se recojan y sean accesibles al conjunto de la ciudadanía, organizaciones y profesionales interesados, los informes de evaluación de los aspectos clínicos y no clínicos, especialmente los de eficiencia económica y de posicionamiento, así como toda aquella información relevante utilizada en el proceso de ETS que no incurra en cuestiones de confidencialidad según la regulación vigente al respecto.
Justificación: Es fundamental que la sociedad conozca el los precios y costes de las tecnologías sanitarías, así como los beneficios que aporta y su nivel de evidencia tras un proceso de comparación con las tecnologías existentes. Esto supone un paso en la democratización y corresponsabilidad en las tomas de decisiones que influyen en la sostenibilidad del SNS, así como la oportunidad de abrir nuevos ámbitos de estudio e investigación.
65. Enmienda de adición nueva .Disposición adicional segunda.
El Ministerio de Sanidad, junto las partes del sistema de ETS adoptará una definición de valor añadido de las nuevas tecnologías sanitarias coherente con los objetivos del RD y, en especial, con el valor terapéutico añadido, sus costes de desarrollo y su impacto económico en el SNS.
Justificación: Cada vez son más los ensayos clínicos que recurren a criterios de valoración indirectos o a parámetros no clínicos que predicen una posibilidad razonable de que pueda haber un beneficio. No hay un requisito de eficacia sustancial, lo que deja en manos de clínicos y pacientes suponer el valor terapéutico del medicamento. Por eso, una descripción clara de los efectos beneficiosos es una información esencial para la toma de decisiones. La ETS persigue identificar el valor añadido de las tecnologías sanitarias, sin embargo, no existe una definición consensuada y alineada con todos los parámetros y objetivos a tener necesariamente en cuenta en el proceso para su concreción.
66. Enmienda de adición nueva. Disposición adicional tercera.
El Ministerio de Sanidad revisará, en el plazo máximo de un año tras la entrada en vigor del presente RD, el sistema de aportaciones por volumen de ventas al Sistema Nacional de Salud recogido en la disposición adicional sexta del Real Decreto Legislativo 1/2015, de 24 de julio, por el que se aprueba el texto refundido de la Ley de garantías y uso racional de los medicamentos y productos sanitarios para adecuarlo a éste otros nuevos retos de financiación del SNS, de forma que contribuya de forma suficiente, adecuada y coherente con las obligaciones que recoge la adopción de nuevas normativas en el ámbito nacional y europeo, a la financiación de servicios sanitarios y sostenibilidad de los SNS. Este sistema revisado debe garantizar la financiación de una plataforma pública de I+D que dé respuesta asequible y accesible a los retos en salud, especialmente a aquellos que donde la industria presenta menor interés y/o aquellos en los que su potencial impacto presupuestario así lo aconsejan.
Justificación: el sector farmacéutico europeo representa el sector industrial más competitivo de la UE, con un margen de beneficios Miembros o del doble que la media del resto de sectores industriales europeos, al mismo tiempo que la factura farmacéutica de los sistemas de salud de los Estados Miembros supone un obstáculo a la sostenibilidad de los mismos, representando de media un 20% de sus presupuestos sanitarios y en torno al 1,6% del PIB de los países de la UE. Además, las nuevas tecnologías no sólo facilitarán el impulso de la I+D, sino que también reducirán sus costes. Finalmente, la financiación pública de la I+D representa más del 40% respecto a la privada hoy en día. En definitiva, este nuevo sistema debe responder hacia una adecuada proporcionalidad y coherencia con las ganancias y margen de beneficios medio de los desarrolladores y, en todo caso, garantizar la asequibilidad y accesibilidad de la tecnología sanitaria, sobre todo la que ha recibido financiación y/o apoyo público.
- La Asociación por un Acceso Justo al Medicamento es una entidad de carácter voluntario y sin ánimo de lucro sometida a la legislación vigente, en particular, a lo dispuesto en el art. 22 de la Constitución Española desarrollado por la Ley Orgánica 1/2002, de 22 de marzo, reguladora del derecho de asociación y normas complementarias, así como a por sus Estatutos.
- Justificación y motivos.
La Organización Mundial de la Salud, considera que el acceso equitativo a unos medicamentos seguros y asequibles es de una importancia vital para que la población goce del grado máximo de salud que se pueda lograr. El acceso a los medicamentos puede ser una cuestión de vida o muerte, pero no solamente esto, ya que también es fundamental para mejorar la calidad de vida y es primordial para tener una vida digna.
Son muchos los factores que contribuyen a la falta de acceso a los medicamentos: la pobreza, no disponer de sistemas públicos de salud de cobertura universal y el uso abusivo de las leyes de propiedad intelectual o patentes de medicamentos.
El gravísimo problema de la falta de acceso a los medicamentos, no es exclusivo de los países pobres o en vías de desarrollo, sino que también ha llegado ya a los denominados países desarrollados.
Estamos asistiendo en la actualidad a una presión insoportable de algunas compañías farmacéuticas, debido a su posición domínate en base a las patentes otorgadas por los gobiernos, que están rompiendo los equilibrios tradicionales al poner unos precios injustificados y abusivos. Esta presión está poniendo en riesgo la sostenibilidad de nuestro Sistema Nacional de Salud y obstaculizando el acceso de las poblaciones más vulnerables a los nuevos medicamentos. El Sistema Nacional de Salud tiene que detraer recursos de otras necesidades de atención sanitaria, del gasto en otro tipo de servicios y del gasto en remuneraciones del personal y de la incorporación de nuevos profesionales.
El abuso de la patente por parte de las compañías farmacéuticas al fijar precios altísimos y desproporcionados hace que la barrera de acceso sea el precio y no el coste. La dificultad de financiación de estos medicamentos no está en el coste de la fabricación, ni en el coste de la investigación, si no en el precio que ponen los fabricantes que llega a ser 20,30 y más de 100 veces superior del coste de fabricación y de investigación.
El precio del medicamento debe cubrir el coste de producción y cubrir la inversión realizada en Investigación más Desarrollo, y permitir un beneficio razonable, que no debe exceder del 10%.
Líneas de trabajo.
• Sensibilización a profesionales, al conjunto de la sociedad. Preparar presentaciones: entrevistas, charlas, conferencias.
• Monitorizar problemas de Acceso a los medicamentos. Dimensionar y monitorizar el problema. Estadísticas, encuestas. Testimonios de personas que no pueden comprar los medicamentos. Servicios de salud que no pueden comprar por precios altos, retirada de medicamentos baratos, etc. Promover Observatorio. Elaborar Informe semestral con índice de accesibilidad.
• Monitorizar la evolución de los costes, los precios y las ganancias de los laboratorios. Para poder objetivar si hay un abuso de patente, un abuso de posición dominante por precios no equitativos se debe conocer cuánto cuesta el medicamento (fabricación e I+D), cuál es el precio que se paga, y qué beneficios obtienen las empresas.
• Estudio de la aplicación de las patentes de medicamento y de otros instrumentos de protección de la Propiedad Intelectual (Certificado de protección suplementaria, exclusividad de datos, etc.). Seguimiento de normativa en España y en la UE. Tratados internacionales.
• Analizar y tratar de objetivar qué es abuso de posición dominante por precios no equitativos. Proponer las modificaciones legales, convenios internacionales, etc., que tiendan a prevenir y corregir el abuso del monopolio creado con los instrumentos de protección de la Propiedad Intelectual.
• Licencias Obligatorias: Analizar con las Consejerías y el Ministerio, con otros países, organizaciones y expertos la aplicación de Licencias Obligatorias: dificultades, estrategias, etc. Preparar proyectos de normativa para hacer más fácil y segura la aplicación de licencias obligatorias cuando sea preciso.