Los costes de producción y de investigación y desarrollo tecnológico deben de ser el criterio principal para la fijación de precios de los medicamentos para que su coste sea calculado en función de su coste real, de manera objetiva y transparente.
Así lo considera la Asociación por un Acceso Justo al Medicamento (AAJM) en sus propuestas enviadas hoy al Ministerio de Sanidad para el proyecto de Real Decreto por el que se regulan los procedimientos de financiación y precio de los medicamentos, cuya consulta pública finaliza hoy, 15 de enero.
Este numero, correspondiente a los meses de noviembre y diciembre de 2024, arranca deseando a todos un feliz cambio anual de ciclo y expresando de deseo de conseguir el derecho efectivo e universal a la salud.
El editorial, de Fernando Lamata, presidente de la Comisión Editorial de la rAJM, analiza la negociación de Tratado de Pandemias, promovido por la OMS, en el que aprecia la pugna entre codicia y equidad y subraya la necesidad de “presionar desde el otro lado. Desde la conciencia colectiva del conjunto de la sociedad y de los pueblos. Frente a los cien mil millonarios, estamos todos los demás, la sociedad de la gente de a pie, el movimiento ciudadano en sus diversas formas”.
Entre los originales, por ser un tema eminentemente propio, destaca el acto de entrega de los premios AAJM 2024, que se celebró el 23 de noviembre con la excelente hospitalidad del Ayuntamiento de Noblejas. Joan Ramon Laporte, Mercedes Zurita, AVITE, Oriol Güell y NoGracias, fueron los galardonados y Manuel Rico (Investigate Europe), tuvo un reconocimiento especial. Se recogen los méritos de cada uno y las declaraciones de los mismos enlazadas a vídeos en YouTube. Asimismo, Soledad Cabezón, presidenta de la AAJM, aprovechó la ocasión para hacer un discurso muy didáctico en el que desmenuzó como la actual política farmacéutica mata y atenta contra el Derecho Humano a la salud.
Entre los restantes originales, igualmente oportunos y adaptados al análisis crítico de la situación actual, se cuenta con el trabajo de Félix Bermejo Pareja y María Victoria Zunzunegui, profesor emérito de CIBERNED el primero y epidemióloga la segunda, revisan las terapias en la enfermedad de Alzheimer y la aprobación de lecanemad y concluyen indicando que “los datos recientes indican una disminución del riesgo de demencia del anciano (y EA) apoyan la necesidad de una estrategia de prevención durante toda la trayectoria vital mediante una acción holística, médica y social y opuesta a la aprobación y acceso universal a tratamientos farmacológicos de dudosa eficacia y seguridad”. El siguiente original de Antonio Pujol de Castro, médico residente de Medicina Preventiva y Salud Publica, vocal de la Comisión Nacional de la Especialidad, de la Red Europea de Residentes de Salud Pública y de la AAJM, abre un debate con el artículo sobre vacunas publicado en el anterior número de la revista por Juan Gervás y Mercedes Pérez-Fernández. Desde el punto de vista de Pujol hay que denunciar los precios abusivos y “no perder en nuestro discurso el abogar por sistemas productivos públicos que suministren y den respuesta a las necesidades reales de la población y por prestaciones sociales suficientes”
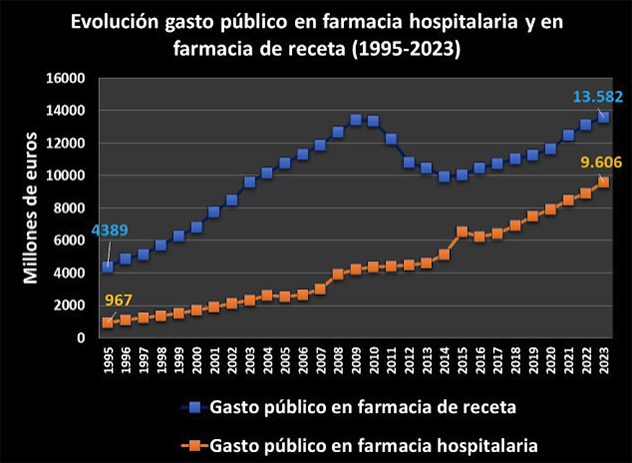
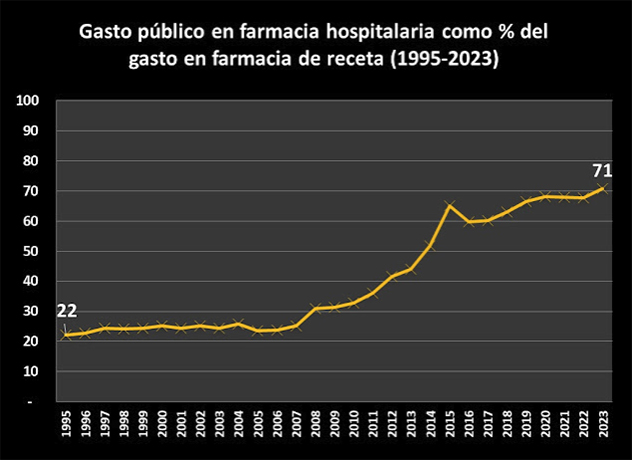
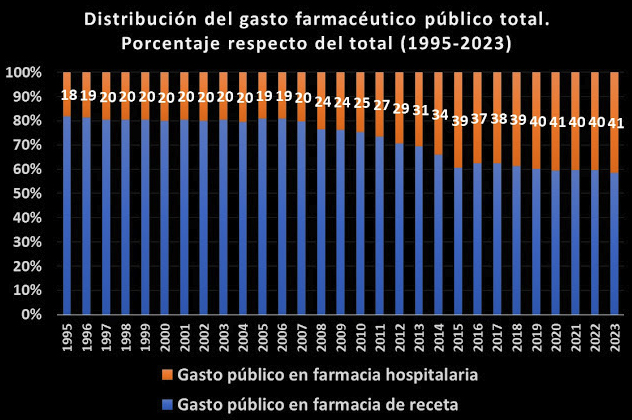
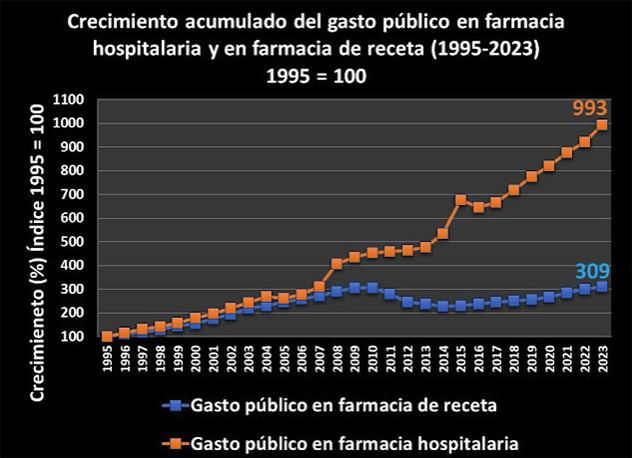
En otras fuentes, el Nº 34 de la rAJM, recoge la conferencia de Fernando Lamata sobre aspectos éticos de la investigación celebrada en el Instituto de Salud Carlos III; el preciso análisis cuantitativo de Juan Simó sobre el gasto público en farmacia hospitalaria, que se ha multiplicado por 10 en 28 años; la valoración de Make Medicinas Affordable a partir del anuncio de Gilead sobre su licencia voluntaria para lenacapavir, que les lleva señalar que “el mundo no puede confiar en las empresas farmacéuticas para garantizar el acceso a los medicamentos esenciales”, y, finalmente, la reflexión de Dean Baker, en la que desarrolla su idea de financiación pública directa de la investigación, para de esta manera conseguir la reducción total de los precios abusivos generados por el sistema actual de investigación de nuevos fármacos de las farmacéuticas basado en las patentes y el monopolio.
Estas son fechas en el año de cambio de ciclo. Las horas de luz solar que se han ido acortando cada día invierten su tendencia y comienzan a alargarse. Es el solsticio de invierno, el instante en el que la Tierra en su órbita elíptica alrededor del Sol se encuentra a mayor distancia. Desde el Neolítico; es decir, desde el inicio de la agricultura, las civilizaciones asiáticas y grecorromanas tuvieron una concepción cíclica del tiempo derivada de la observación de la salida y la puesta del Sol, las fases de la Luna, los movimientos anuales de las estrellas, la sucesión de las estaciones… Una idea del tiempo cíclico que cambiaron las religiones monoteístas descendientes de Abraham (judaísmo, cristianismo e islam), que consideraron predominante el tiempo lineal, el que puede compararse con el transcurso de la vida (nacimiento, desarrollo y muerte). Desde el punto de visto científico, el universo está en expansión acelerada desde el Big Bang y, por tanto, el tiempo es lineal (1).
No obstante, la celebración del solsticio de invierno sigue presente y puede ser considerada la fiesta social más antigua. Se celebra desde hace más de 4.700 años. Primero en Sumeria con Dumuzi, que pasó a Sacaera en Babilonia, Kronia en Grecia y las Saturnales romanas. El 25 de diciembre, día del Natalis Solis Invicti se consideraba la fecha del nacimiento de Apolo y, asimismo, de Mitra y de Buda.
Buscando el sincretismo y muy especialmente contrarrestar el culto a Mitra que también era popular en Roma en el siglo IV de nuestra era, el Papa Julio I, en el año 337, estableció que la fecha del nacimiento de Jesucristo se celebrara el 25 de diciembre, el mismo día del Sol Invicto. Este es el origen de la actual tradición cristiana (2).
Desde el respeto …. a todas las creencias, y a la articulación de narrativas ajustadas a la interpretación de la realidad de cada uno, que muy seguramente será real en sus consecuencias, buscando un objetivo común de celebración en el cambio de ciclo, permítanos aprovechar el solsticio de invierno para desearles felicidad ahora y ventura en los meses venideros, con un acceso justo a los medicamentos, que permita conseguir para toda la humanidad el derecho a la salud.
Finalmente, un breve poema dedicado a Valencia y su Amanecer
EDITORIAL. Revista nº 34 Noviembre – Diciembre 2024
Fernando Lamata.
Presidente de la Comisión Editorial de la rAJM.
Entre el 4 y el 15 de noviembre pasado se celebró la décimo segunda sesión del Ente Negociador Internacional (INB en sus siglas en inglés) para el Tratado de Pandemias promovido por la OMS. No se logró un acuerdo, por lo que el INB seguirá negociando en los primeros meses de 2025 para intentar alcanzarlo en la Asamblea Mundial de la Salud en 2025. Recordemos que fue esta Asamblea Mundial la que decidió hace más de tres años impulsar este Tratado en su reunión de mayo de 2021.
A la vista del último borrador del INB, podemos decir que el Tratado no cumplirá los objetivos que pretendía. De hecho, ya no se atreven a llamarlo Tratado, sino “Convención, Acuerdo u otro instrumento internacional…”. En cambio, la presión de las grandes empresas farmacéuticas y de sus asociaciones sobre los gobiernos de los países de altos ingresos está logrando su propósito: mantener como intocables los Derechos de Propiedad Intelectual (DPI) en los medicamentos y productos sanitarios.
En los momentos más duros de la pandemia de la COVID-19, África y la India, junto con otros países del Sur Global, plantearon en la Organización Mundial del Comercio (OMC) la necesidad de aplicar una exención temporal de los Derechos de Propiedad Intelectual para todos los productos relacionados con la pandemia (vacunas, diagnósticos, tratamientos) mientras durara la misma. ¿Por qué lo demandaban? Porque estos Derechos de Propiedad Intelectual, patentes y otras exclusividades confieren a las empresas titulares el monopolio sobre el producto. Y, al tener el monopolio, son las empresas las que deciden cuánto producen, dónde producen, a quién venden y a qué precio venden. Recordemos que, durante la pandemia de la COVID-19, los gobiernos patrocinaron la investigación y desarrollo de vacunas, diagnósticos y tratamientos; pero cedieron los Derechos de Propiedad Intelectual a las empresas y, como consecuencia, hubo retrasos en la disponibilidad global de vacunas y otros productos, mientras se acaparaban en los países ricos, que tuvieron que pagar 10 y 20 veces por encima de los costes de producción, generando enormes ganancias abusivas a las empresas productoras. Al mismo tiempo, por el impacto de la pandemia murieron más de 20 millones de personas en el mundo y muchas otras enfermaron y sufrieron (todavía sufren) importantes secuelas; la economía se vio afectada de forma severa y se destruyeron millones de puestos de trabajo. Entonces, las empresas farmacéuticas y sus lobistas, a través de los representantes de EEUU y la UE, frenaron en la OMC la exención de los Derechos de Propiedad Intelectual. El Tratado era una segunda oportunidad para que los países pusieran los derechos humanos de todos por delante de las ganancias abusivas de unos pocos. No parece que vaya a ser así.
El último borrador del Tratado (de 14 de noviembre, a las 20.00), reconoce en su preámbulo que la protección de la propiedad intelectual es importante para el desarrollo de nuevos medicamentos (1). Recoge así la narrativa de la industria farmacéutica, pero no afirma la verdad: los monopolios generados por los Derechos de Propiedad Intelectual en medicamentos matan (no solo en pandemias), y no promueven el desarrollo de nuevos medicamentos. En efecto, la mayor parte de la investigación innovadora es de financiación pública directa. Los monopolios generan enormes beneficios por sobre precios, que supuestamente debían ir a I+D, pero van en su mayor parte a marketing y a remuneración de los altos ejecutivos y accionistas. Los precios abusivos suponen una barrera al acceso a los medicamentos y generan una serie de efectos secundarios adversos (sesgos de investigación, sobre prescripción innecesaria y nociva, déficit público, etc.). Y, sin embargo, los países de altos ingresos siguen cediendo ante la presión del lobby de la industria. Así, el Tratado no habla de que durante las pandemias se aplicará una exención de los Derechos de Propiedad Intelectual, sino que se incluyen referencias a las flexibilidades del Acuerdo sobre los Aspectos de los Derechos de Propiedad Intelectual relacionados con el Comercio, que permiten a los países, entre otras posibilidades, aprobar Licencias Obligatorias (autorizando la fabricación o importación de genéricos y biosimilares, aunque exista patente). Pero esa herramienta ya la tienen. El Tratado debería ir más allá, estableciendo un automatismo en la exención de los DPI y evitando así la presión de la Big Pharma sobre los países para que no apliquen dichas Licencias Obligatorias u otras medidas similares.
El artículo 11 del borrador del Tratado se refiere a la Transferencia de Tecnología y Conocimiento para la producción de productos sanitarios relacionados con la pandemia. Este es un punto clave, que ha subrayado la organización Knowledge Ecology International (KEI) (2). Los lobbies de Big Pharma han presionado para que el texto se refiera a la Transferencia de Tecnología como un proceso voluntario, consensuado, no obligatorio. KEI propuso una redacción alternativa dejando claro que la ley nacional puede imponer la transferencia de tecnología en caso de necesidad. En otros apartados del borrador, que siguen en discusión, se indica que los productos y tecnologías de propiedad pública se deberían ofrecer con licencias no exclusivas a países de bajos ingresos. Así mismo, en el Artículo 9.5 se indica que en investigación con patrocinio público se deberían establecer licencias no exclusivas, transferencia de tecnología y precios asequibles. Son aspectos positivos, pero insuficientes. Sería preferible que el Tratado estableciera como obligatoria la Transferencia de Tecnología y Conocimiento, en caso de pandemia, con carácter general.
La Transferencia de Tecnología y Conocimiento es clave ya que permitiría la fabricación en las diferentes regiones del planeta, en países de diferente nivel de desarrollo, y posibilitaría la venta de los productos a precios de coste de producción, logrando que el acceso a estos productos fuera equitativo en todo el mundo.
En este sentido, el Artículo 10 habla de fomentar la diversificación de la producción, lo cual está bien. Pero crear o reforzar esos nodos de producción requiere financiación para que pueda hacerse en países de bajos ingresos, lo que no está garantizado en el Tratado, y requiere también, como hemos visto, la transferencia de tecnología y conocimiento, que tampoco están garantizadas en el Tratado.
Otro aspecto en discusión es el sistema de Acceso a Patógenos y Beneficios Compartidos (PABS system) que recoge el artículo 12. Los países de altos ingresos insisten en la primera parte del sistema, que se refiere a compartir de forma inmediata los datos sobre brotes, los patógenos causales, etc. y a la toma de medidas para frenar los brotes iniciales. En cuanto a la segunda parte, compartir los beneficios, ya se ha visto que no aceptan la exención de los Derechos de Propiedad Intelectual, con lo cual, el acceso a vacunas y otros productos dependerá de cuánto y dónde quieran producir las empresas y a quiénes y a qué precio quieran vender. Nada de esto se regula. En el borrador actual se incluye una mención a que se deberá ceder el 20% de la producción a la OMS, la mitad gratis, y la mitad a precio de coste, para distribuirla en función de los riesgos para la salud pública y la necesidad. Sería, desde luego, un avance. Pero los países de baja renta, con razón, insisten en que este mecanismo sigue siendo caridad y no el reconocimiento de un derecho.
En el Artículo 13 se crea una red de cadenas de suministro y logística global (GSCL), que organizaría la OMS y que deberá procurar el acceso equitativo, asequible y universal a los productos sanitarios relacionados con la pandemia. Esta herramienta, en una OMS con autoridad reforzada por delegación de los países, podría jugar un papel importante (en vez del mecanismo ACT Accelerator-COVAX, controlado por las empresas, durante la pandemia de la COVID-19) (3). En un Artículo 13 bis se insiste en la transparencia en los mecanismos de compra y distribución, así como en las condicionalidades que faciliten un acceso equitativo y universal. Serían pasos positivos.
Por el contrario, un flanco débil del Tratado es la financiación. Se habla de un Mecanismo de Coordinación Financiera con contribuciones de carácter voluntario. Los países de bajos ingresos, en cambio, reclaman una financiación adicional, que debería venir de aportaciones obligatorias en proporción a la renta de cada país. Sin financiación adicional no podrán cumplir sus compromisos (reforzar los sistemas de salud, detección y respuesta temprana a los brotes, sistemas de información, generar instalaciones para la producción local de productos sanitarios, etc.). Para Wemos sería conveniente que el Tratado citara que esa financiación adicional debe venir de sistemas fiscales progresivos y apoyara la Convención de Naciones Unidas sobre marco de cooperación fiscal, que está en discusión (4). Otra fuente de financiación que reclaman los países de bajos ingresos es la reestructuración de la deuda, la suspensión del servicio de la deuda o la cancelación total o parcial de la misma. Esta posibilidad se mencionaba en versiones anteriores y ha desaparecido.
En su actual redacción, el Tratado tendría otros aspectos positivos como reforzar el papel de la OMS en su papel de organismo multilateral y recordar los valores de la equidad, la solidaridad y la sanidad universal. Pero no evitaría que una futura pandemia fuera, otra vez, una catástrofe, porque no garantiza, de forma obligatoria y automática, la exención de patentes, la transferencia de tecnología y conocimiento, la producción distribuida de forma equitativa y precios a coste de fabricación(5).
Noam Chomsky publicó en 2016 su libro “¿Quién domina el mundo?”. Lamentablemente, cada vez más, lo dominan unos pocos cien mil millonarios y unas pocas corporaciones multinacionales, con enorme poder sobre los Gobiernos y los Parlamentos nacionales. En la reunión del G-20 celebrada esta semana en Brasil, los líderes políticos han coincidido en que para hacer frente a los desafíos mundiales es preciso reforzar la gobernanza política global y desarrollar soluciones multilaterales (6). En efecto, sería preciso reforzar el sistema multilateral, reformando el Consejo de Seguridad, el Consejo Económico y Social y el Secretariado de la ONU, así como el FMI, el Banco Mundial y otras estructuras, en línea con lo propuesto por el profesor Luigi Ferrajoli en la Constitución de la Tierra.
Sin embargo, los superricos no quieren perder el poder que han recuperado en los últimos 30 años. Y por eso promueven iniciativas políticas “anti-políticas”, “anti-sistema”, “anarco-capitalistas” y otras similares para desacreditar a las estructuras políticas y desacreditar los intentos de reforzar la gobernanza política mundial. Por eso financian a líderes nacionalistas, que rechazan a las organizaciones multilaterales, centran sus ataques en las estructuras burocráticas y las regulaciones (las que no favorezcan a los superricos), denigran las políticas contra el cambio climático y el consumo de combustibles fósiles y tratan de construir la imagen del adversario en los inmigrantes, en las feministas, los homosexuales, las personas con discapacidad, las personas mayores, los sindicalistas, etc., según el viejo lema: divide y vencerás.
Una ofensiva mediática y académica, bien financiada por los poderes económicos, refuerza el discurso del neo-capitalismo financiero global. Se fomenta el individualismo, el sálvese quien pueda, la fragmentación social. Lo hemos visto en las inundaciones de la Comunidad Valenciana el 29 de octubre: cuentas de redes sociales con patrocinio oculto e interesado han difundido mensajes para tratar de desacreditar los esfuerzos de las instituciones y los funcionarios públicos, y han resaltado la idea de que “el pueblo salva al pueblo”. Un mensaje antipolítico que, manipulando la imagen ejemplar de miles de voluntarios y de su entrega solidaria, pretende ignorar el trabajo de los bomberos, policías locales, guardias civiles, militares, sanitarios, trabajadores de Adif y de Renfe, trabajadores de obras públicas, etc., etc. Miles de funcionarios y trabajadores pagados con dinero público, que se esfuerzan para recuperar las infraestructuras dañadas, y, por otro lado, miles de ayudas públicas, que saldrán del presupuesto público, es decir, de los impuestos, del Estado, para familias, empresas y ayuntamientos.
En este contexto, llamo la atención sobre la aprobación por el Congreso, el pasado 21 de noviembre, en una sesión malabarista, y por una exigua mayoría, de la propuesta del gobierno de España para intentar mejorar la recaudación pública llevando adelante una serie de reformas fiscales que incluyen el tipo mínimo del 15% a las empresas multinacionales, el impuesto diluido por la presión de los lobistas) a los beneficios de la banca y a las eléctricas, así como a los vapeadores y las labores del tabaco, o el aumento del IRPF para las rentas del capital (7). Conviene recordar que sin impuestos justos y progresivos (donde pague más el que más tiene) no puede haber servicios públicos de calidad.
También conviene tener claro que hacer frente a los grandes retos que nos amenazan, como el cambio climático y su efecto devastador en catástrofes naturales, las guerras, las desigualdades económicas crecientes, y también las pandemias, tenemos que trabajar juntos en desde el multilateralismo democrático.
El Tratado de Pandemias recoge buenas ideas, pero no refuerza la OMS de forma suficiente (financiación adecuada a los retos). Y no cambia el discurso y las reglas sobre los Derechos de Propiedad Intelectual y los monopolios, ni sobre la Transferencia de Tecnología obligatoria y automática en caso de pandemia. De esta forma es difícil que podamos responder a una nueva emergencia de salud internacional con medicamentos y vacunas eficaces, en volumen suficiente, fabricados en todas las regiones del mundo, distribuidos de forma equitativa, según necesidad, y comercializados a precio de coste. Y si no lo hacemos así, en una pandemia de alta contagiosidad y letalidad, toda la humanidad sufrirá las consecuencias.
Los gobiernos de los países de altos ingresos, EEUU y la UE, no parecen reaccionar, influidos por los intereses y la narrativa de la Big Pharma. Con estas inercias, el Tratado que se apruebe en mayo de 2025 será un paso muy débil. ¿Cómo cambiar esta situación? Presionando desde el otro lado. Desde la conciencia colectiva del conjunto de la sociedad y de los pueblos. Frente a los cien mil millonarios, estamos todos los demás, la sociedad de la gente de a pie, el movimiento ciudadano en sus diversas formas de asociación, las organizaciones sindicales, las organizaciones de consumidores y otras muchas: las organizaciones de la sociedad civil. Se está enfrentando la codicia frente a la equidad. Tenemos que ser capaces de ver que la codicia insaciable de unos pocos nos está llevando al precipicio: el 1% de los más ricos tienen tanta riqueza como el 95% de la población. Es un abuso que afecta a nuestras posibilidades de supervivencia. El Tratado de Pandemias es una ocasión de revertir esta expropiación sistemática que hacen unos pocos de los bienes públicos que son de todos. Es una oportunidad de mostrar el camino para una gobernanza global reforzada que permita afrontar los retos del presente y del futuro. En estos meses debemos seguir insistiendo. Nos queda la palabra, la voluntad y la esperanza.
Referencias
(1). Borrador del Tratado de Pandemias. 14 noviembre 2024, 20.00
La aprobación de lecanemab en EEUU como terapia de la enfermedad de Alzheimer (EA) y su no aprobación por la EMA (European Medical Agency) ha suscitado discusión. Presentamos un análisis crítico de esta terapia y una breve revisión nosológica de la EA.
Material y Métodos
Revisión de autores, con búsqueda de la literatura en bases biomédicas, fundamentalmente, en MEDLINE y Google Académico.
Resultados
Se efectúa una sucinta revisión de las terapias farmacológicas de la EA, y se analiza con más detalle la que considera que la fisiopatología de EA (hipótesis amiloide) está determinada por un depósito cerebral excesivo de β-amiloide, que requiere su eliminación mediante inmunización activa (vacunas) o pasiva (anticuerpos humanizados anti-β-amiloide). Estas estrategias no han tenido éxito (no evitan su progresión) y producen mínima (o dudosa) mejoría clínica no exenta de efectos adversos. Se analiza cómo el lecanemab, exponente de estos anticuerpos, no ha sido aprobado en Europa. Se cuestiona esta nosología sobre la EA por su fracaso terapéutico y se discute la asociación EA-envejecimiento y la reciente disminución de riesgo de demencia (y EA) en países ricos lo que apoya su prevención mediante intervenciones para reducir los factores de riesgo en cada etapa de la trayectoria vital.
Conclusiones
La no aprobación del lecanemab por la EMA se juzga positiva. Se requieren más estudios sobre las asociaciones: EA-envejecimiento y EA de múltiple patología. La disminución del riesgo de demencia en países ricos apoya la prioridad de su prevención frente a tratamientos farmacológicos de dudosa eficacia y seguridad.
Texto
1.) Introducción
Antes de comentar la terapia de la enfermedad de Alzheimer (EA) conviene recordar algunos puntos conceptuales e históricos de esta enfermedad.
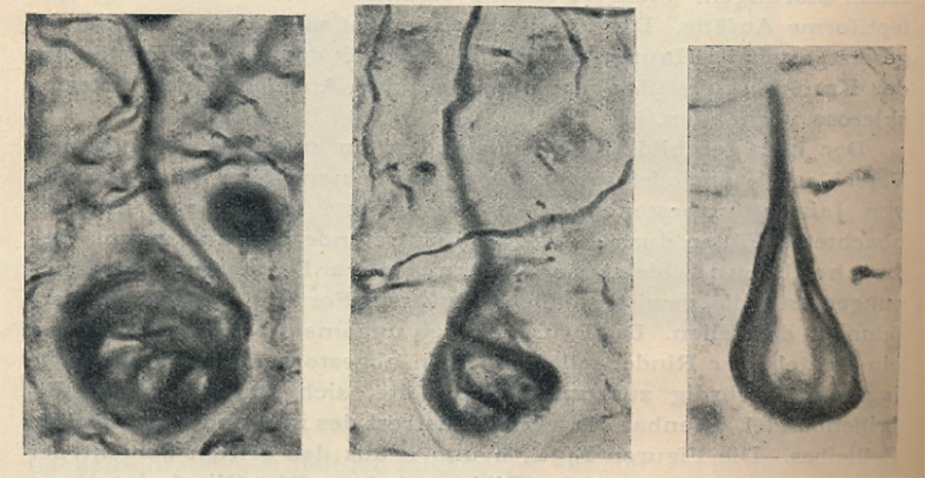
La (EA) nació como premio del entonces más prestigioso psiquiatra del mundo, Emil Kraepelin, a uno de sus más brillantes discípulos, Alois Alzheimer, con una nueva enfermedad que llevaría este epónimo, EA, y con él la introdujo en su texto de psiquiatría de 1910 (Kraepelin, 1910) con muy pocos casos descritos. La nueva enfermedad era una demencia presenil con cambios histológicos peculiares, los ovillos neurofibrilares o degeneración neurofibrilar (DNF, acrónimo inglés), descritos por Alois Alzheimer en 1906; anteriormente esta demencia se asociaba con las placas seniles cerebrales (Alzheimer, 2011; Assal, 2019). Véase las figuras 1 y 2 en las que se representa las dos lesiones más características de esta enfermedad, aunque otras importantes son la pérdida neuronal y sináptica (Jellinger et al, 2020).
Figura 1. Ovillos neurofibrilares (NFT) en la enfermedad de Alzheimer
Imágenes de tres neuronas con ovillos neurofibrilares (o degeneración neurofibrilar –NDF) en su interior (ovillos de hilos gruesos en negro en el soma neuronal).
Foto reproducida del libro de Kraepelin de 1910 (véase Kraepelin, en la bibliografía)
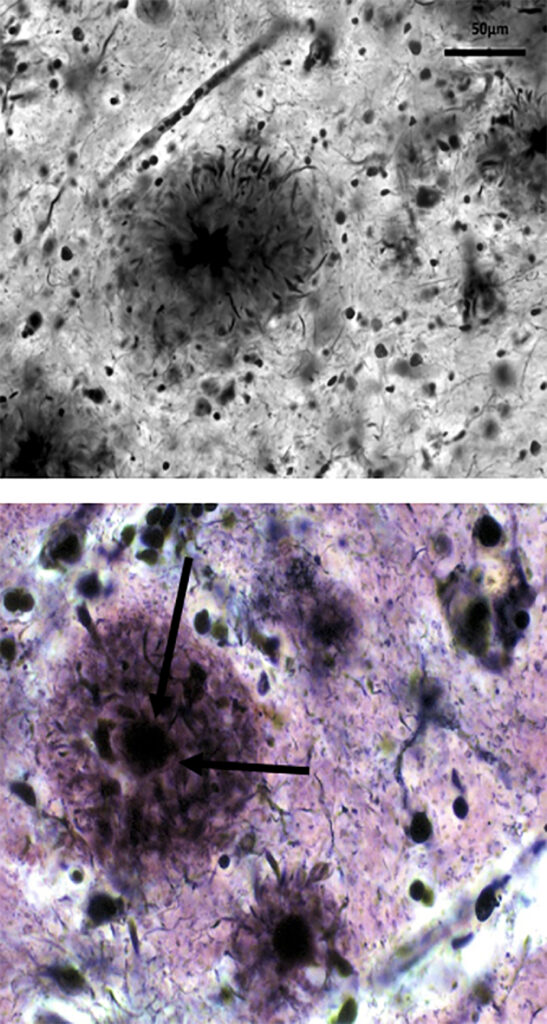
Figura 2. Placas seniles neuríticas en la enfermedad de Alzheimer
Imágenes de las placas seniles neuríticas. En la imagen superior se observa la placa senil (imagen redondeada en negro en el centro de la imagen) y se ven las neuritas alteradas que la rodean. Y en la foto inferior se visualiza muy bien en núcleo central de la placa (depósito de beta-amiloide), como indican las dos flechas, rodeada de neuritas.
Las imágenes pertenecen al archivo Cajal y han sido donadas por el Dr. Martínez, ex director del Instituto Cajal, para su reproducción no comercial.
(Las exponemos porque la calidad de las imágenes es superior a la de otros muchos neuropatólogos coetáneos).
Los coetáneos a Alois Alzheimer consideraron, la EA, una forma muy infrecuente de demencia presenil, pero ésta generó desde su nacimiento una indudable controversia de gran interés histórico y científico (Amaducci, 1996; Bermejo-Pareja y del Ser, 2024), que está fuera de la intención de este trabajo.
Berchold y Cotman (1998), examinando la historia de la demencia, comentan que desde la descripción de la EA hasta 1960 la atención médica a esta enfermedad fue casi puramente clínico-patológica, siendo el interés primordial discutir si la EA era o no una forma diferente de demencia senil.
Tras la segunda guerra mundial, el panorama de la demencia cambió considerablemente, sobre todo en EEUU: el envejecimiento poblacional, el incremento de su longevidad y su nuevo enriquecimiento en la ya afluente sociedad americana, determinó una influencia cada vez mayor de este grupo etario en todos los órdenes sociales. Según el historiador de la ciencia, Ballenger (2006), la sociedad americana aceptó la propuesta de diversas instancias médicas de la medicalización del declive cognitivo y de la demencia en la ancianidad, que había sido considerada “natural” durante siglos, convirtiéndola en una enfermedad. Tres brillantes médicos e intelectuales fueron los artífices de este cambio, Robert Butler, psiquiatra, premio Pulitzer, luchador contra el “ageism” (o edadismo, en su traducción en español) (Butler, 1969), y primer director del National Institute on Aging (NIA) norteamericano (Achenbaum, 2014). Otro gigante al que corresponde un gran mérito fue Robert Katzman, neurólogo e ideólogo de “changing view” (Katzman y Bick, 2000), que reconvirtió una rara demencia presenil en la demencia presenil y senil, cuya suma la hizo más frecuente, tanto que se convirtió en una causa mayor de mortalidad de los mayores en EEUU (Katzman, 1976; Kawas, 2009). A ellos se sumó un prestigioso neuropatólogo, Robert Terry, que le dio carta de naturaleza patológica a lo largo de décadas, aunque él consideraba que la causa verdadera de EA era un déficit sináptico (Terry RD et al, 2009 y Terry NA et al, 2019). En ese contexto histórico, el NIA consideró el tratamiento y curación de esta enfermedad como uno de los primus movens de su quehacer. El contexto científico europeo y mundial fue aceptando esta proposición poco a poco como demuestran varios estudios bibliométricos (Serrano-Pozo et al, 2017; Robert et al, 2020). Muy pocos médicos criticaron, entonces, la restringida postura del NIA, dejando en un lugar secundario la investigación del envejecimiento (Adelman, 1998).
Con el empuje teórico y económico del NIA y con la intervención entusiasta de la industria farmacéutica se han generado desde entonces múltiples intentos terapéuticos.
2.) Terapia farmacológica en el EA. Perspectiva histórica, y el caso del lecanemab
El NIA, siguiendo su política en investigación y “cura” de la EA le ha dedicado ingentes cantidades de dinero procedentes de este Instituto Federal americano (Fox, 1989), y posteriormente también en EEU, la Asociación de Alzheimer (última denominación de este lobby de apoyo a los enfermos de EA), y en general, en todo el mundo. Y ha habido logros indudables, como ejemplo el hallazgo de la degeneración de las neuronas colinérgicas del tronco cerebral descubierto por Whitehouse et al (1981), que sustentó morfológicamente el déficit de inervación colinérgica de la corteza cerebral en la EA, que, asociado a la previa verificación de la importancia de esta inervación con la memoria (Drachman y Leavitt, 1974; Bartus et al, 1982), permitió el desarrollo de fármacos colinomiméticos (aumentan la estimulación colinérgica), fundamentalmente inhibidores de la colinesterasa (fisostigmina, donepezilo, rivastigmina y otros) que han entrado en el arsenal terapéutico con éxitos pírricos en los ensayos clínicos, pero muy limitados en la práctica médica, pues solo producen mejoría clínica en un porcentaje reducido de casos, cuyo beneficio no suele superar dos años, y no desdeñables efectos secundarios, y desde luego, no curan ni el deterioro cognitivo progresivo de la EA ni atenúan su mortalidad (Sharma, 2019; Bermejo-Pareja y del Ser, 2024). Hay visiones algo más optimistas desde la perspectiva fisiopatológica de estos fármacos (Hampel et al, 2018).
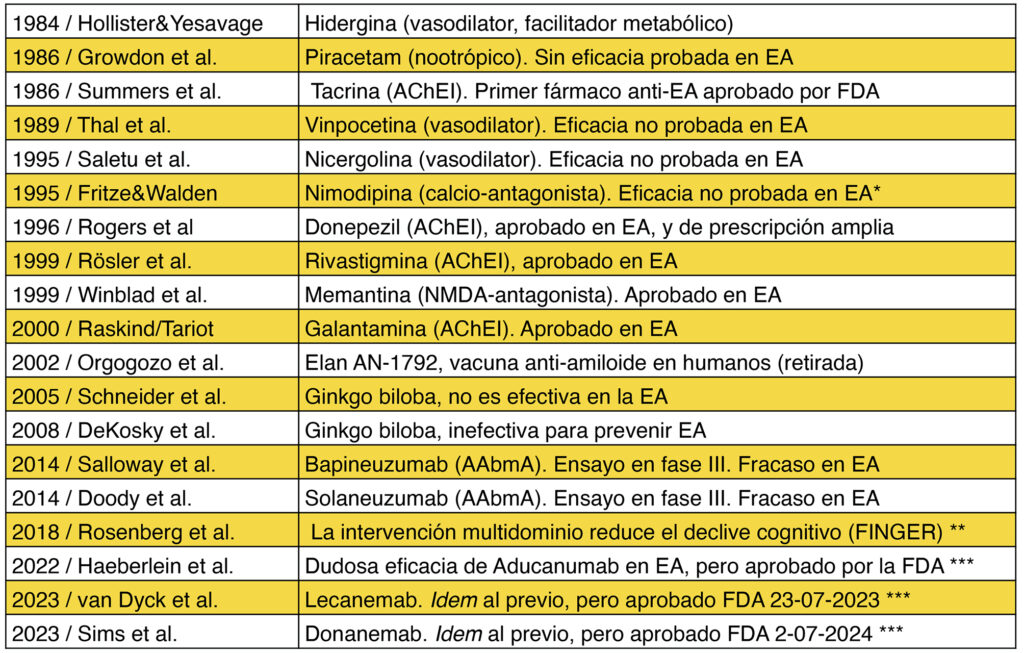
El ingente desarrollo de fármacos contra la demencia y la EA (en la tabla 1 se expone una mínima muestra) tiene una línea preeminente que proviene de la genética, cuyo brillante desembarco en el siglo XXI, ha descrito nuestro acervo genético (ADN) y entrevisto la posibilidad de actuar en el metabolismo molecular de nuestro organismo. En el ámbito de la EA existen raros casos genéticos de EA familiar (1-5%), pues la mayoría (95-99%) son esporádicos (Bermejo-Pareja y del Ser, 2024). Y sus tres formas genéticas familiares (de comienzo precoz) se deben a alteraciones de los genes de APP, presenilina 1 y 2, cuya consecuencia molecular, en este ámbito, es determinar un exceso de producción de β-amiloide (βA), proteína que se acumula en el “core” central de las placas seniles de la EA (figura 2), y aunque dicho así parece simple, su acción metabólica cerebral es muy compleja (Steiner et al, 1999; Lanoiselée et al 2017; Bertram & Tanzi, 2020;) y generó la hipótesis amiloide (cascada amiloide) de la EA (Hardy & Higgins,1992), que predica, esencialmente, que el acúmulo excesivo de βA es tóxico para el cerebro y determina una cascada amiloide de eventos metabólicos que generan pérdida neuronal y demencia. Sus autores la siguen defendiendo (Selkoe y Hardy, 2016). Sobre esta hipótesis se ha basado la categorización diagnóstica y la investigación farmacológica de la EA durante más de un cuarto de siglo. Se ha intentado con varias estrategias terapéuticas eliminar el depósito excesivo de βA cerebral (Bermejo-Pareja y del Ser, 2024). Para ello se han utilizado modelos de animales transgénicos (Wirak et al, 1991), insertando genes humanos de pacientes con EA que, aunque no reproducen fielmente la EA, han permitido emplear fármacos que eliminan el βA de estos modelos (traspasados a los humanos) mediante inmunogénesis activa (vacunas), y posteriormente, mediante anticuerpos humanizados anti-βA (A-AβA) (Geylis et al, 2006). El bapineuzumab es un ejemplo paradigmático de A-AβA (Doody et al,2014) ¿Cuál ha sido el resultado?: descorazonador. La vacuna contra la EA no entró en el mercado por sus efectos adversos (Orgogozo et al, 2003). Pero el relevante estudio patológico de Nicoll et al (2019), ha mostrado que la inmunización activa se asoció con la desaparición de βA en el cerebro de los pacientes tratados con la vacuna, pero también ha constatado, que esta desaparición no mejoró su demencia que continuó progresando (así como las lesiones tipo tau) y la mayoría murió con demencia avanzada (19 de 22 casos). ¿Y qué ha pasado con los numerosos A-AβA?, pues han tenido un resultado análogo, pese a ensayos clínicos muy extensos y bien ejecutados, tanto en la EA genética (Salloway et al, 2021) como en la EA esporádica. Los A-AβA han demostrado que no alteran el curso progresivo de la demencia ni la mortalidad que esta enfermedad determina, aunque en algunos estudios se han evidenciado mejorías estadísticas en algunos test psicométricos, pero no de clara significación clínica (Foroutan et al, 2019; Ackley, et al, 2021; Lozupone et al, 2024). Y hay revisiones sistemáticas con métodos bayesianos que demuestran que estos fármacos no se han separado de la hipótesis nula (Richard et al, 2021), vaya, que no han demostrado efectos terapéuticos claros en la EA.
¿Y cómo es que se continua con la experimentación de estos fármacos y cómo se sostiene la hipótesis amiloide de la EA?, pues probablemente como se han sostenido muchos paradigmas científicos establecidos durante décadas. Pese a la demostración de una realidad palpable de corrientes críticas poderosas (Joseph et el 2001; Whitehouse y George, 2008; Høilund-Carlsen et al, 2020; Herrup, 2021; Lozupone et al, 2024) los paradigmas científicos son difíciles de desbancar como ha mostrado Kuhn (1970). Y en este caso las críticas han sido significativas (Huang et al, 2019; Nicoll et al, 2019; Mullane y Williams, 2020, Ackley et al, 2021; Kurkinen et al, 2023),y algunas feroces como las del decano de la Universidad de Oxford, AD Smith (2022), que ha llegado a sostener que no era ético continuar con la terapia anti-amiloide en la EA, y que la postura de muchos laboratorios de no dar acceso los ensayos a los investigadores, tampoco lo es. Pero la hipótesis amiloide ha generado una poderosa industria no solo de neurociencia básica y farmacológica sino de pruebas diagnósticas alentadas por la industria farmacéutica y acompañada por una tecnología cara y productiva económicamente, de diversos marcadores de EA (con neuroimagen sofisticada, léase RM, SPECT y PET) y determinaciones en sangre y LCR de las proteínas que no se circunscriben a la investigación médica como sería razonable sino que se utilizan en unidades clínicas más o menos especializadas, y cuyo basamento patológico y valor predictivo es escaso o dudoso (Ritchie et al, 2014; Bermejo-Pareja y del Ser, 2024), pero que mantienen en pie la hipótesis amiloide de la EA con argumentos probablemente más de política médica (mejores diagnósticos para el paciente) y farmacológica empresarial (inversión realizada en este campo), que puramente científica o de valor terapéutico para el paciente con esta enfermedad.
El caso del lecanemab es solo un ejemplo más de otros A-AβA ya utilizados, y que no difiere esencialmente de ellos. Después de la publicación en el New England Journal of Medicine de un único ensayo clínico que demostraba su eficacia en retrasar el deterioro cognitivo en la muestra del estudio (Van Dyck et al, 2023), fue aprobado rápidamente por la FDA (Food and Drug Administration) en julio de 2023 (Hoy, 2023), pese a la polémica suscitada sobre éste y otros estos fármacos semejantes (aducanumab, donanemab) (Mullane y Williams, 2020; Herrup 2021; Pang et al, 2023; Høilund-Carlsen et al, 2024), y hay autores que han dado la bienvenida al fármaco (Schiller et al, 2024), pero otros consideran que es una mala noticia (Kepp et al, 2023; Kurkinen, 2023). Sin embargo, recientemente la EMA (European Medicine Agency) ha rechazado su uso en Europa, alegando que su efecto de retraso del declive cognitivo no contrabalancea los efectos adversos serios asociados a su uso, y por consiguiente que se requieren más ensayos y de más larga duración.
Esta decisión inclina la balanza a favor de los críticos de la terapia con los actuales A-AβA. Y hemos de señalar la enorme atención en la literatura científica que ha determinado este fármaco con más de 350 citas en MEDLINE y, ya, con una revisión sistemática (Chowdhury y Chowdhury, 2023). Nos parece relevante la reacción de varios neurólogos (Burke et al, 2023) en Neurology (revista de la Academia Americana deNeurología), antes su aprobación en EEUU, y es preguntarse: “¿no necesitamos datos suficientes para estar seguros de que no es menos efectivo, mucho más dañino y 100 veces más costoso que el donepezilo?”. Y sostienen que se requieren más estudios y a más largo plazo para su uso. Nuestra opinión es semejante sobre los recientes A-AβA y es que, si el donepezilo no ha cambiado el porvenir de los pacientes con EA, pagar un coste de 100 veces mayor por fármacos no lo van a cambiar no parece una inversión sensata, ni siquiera en sociedades afluentes. No existe un elixir mágico contra la EA, y es muy difícil que lo haya en el contexto actual de conocimientos (Larson, 2014;Khachaturian, 2017) y la terapia anti-amiloide actual no lo es. También, el precursor del lecanemab, el donanemab, ha sido rechazado por NICE, la agencia de medicamentos del Reino Unido (Mahase, 2024). Pero ya hay nuevas estrategias futuras en este campo (Bhadane et al, 2024) y en la terapia de la EA (Zhang et al, 2024).
Tabla 1. Breve resumen de terapias farmacológicas en demencia y EA ¶
Abreviaturas en inglés:
AChEI, inhibidor de la acetilcolinesterasa; NMDA: N-metil-D-aspartato.;
AAbmA: anticuerpo monoclonal anti-amiloide-beta. FDA: Food & Drug Administration de EEUU
*Utilizada en demencia vascular y aprobada para el vasoespasmo por hemorragia subaracnoidea
** El estudio FINGER está realizado en pacientes sin demencia, pero con riesgo de padecerla
*** Estos fármacos reducen la carga de amiloide cerebral, pero pequeña mejoría en la EA según los autores de los estudios (tabla y texto) o muy dudosa para sus críticos (Høilund-Carlsen et al, 2024, y texto). La inmunoterapia activa no mejoró la demencia de la EA en 15 años (Nicoll et al, 2019).
Tomada y modificada de Bermejo-Pareja y del Ser, 2024, (trabajo en el que se citan las referencias de esta tabla y no se repiten en éste para no incrementar su listado).
3.). Demencia y envejecimiento
No es conveniente finalizar este trabajo sobre la EA y su terapia actual con resultados negativos por lo que conviene aclarar algunos puntos sobre este padecimiento. El primero sería sobre su génesis. Si la terapia basada en la hipótesis amiloide no ha conseguido fármacos curativos en más de 25 años ¿No hay otras hipótesis patogenéticas sobre la EA? Sí, hay más de 100, desde un criterio evolucionista (Rapoport, 1989), al infeccioso (Itzhaki et al, 2020), incluso alguno abstruso (Wostyn et al,2010), pero desde una perspectiva bibliométrica, la segunda hipótesis es el envejecimiento (Bermejo-Pareja y del Ser, 2024). Hipótesis que no es ni nueva ni definitiva. El envejecimiento es el más antiguo, estable y mayor factor de riesgo de la demencia, EA y de la mayoría de enfermedades neurodegenerativas (END), pero su relación desde una perspectiva fisiopatológica con estas enfermedades es desconocida (Yakner et al, 2008; Aron et al 2020). Tampoco son bien conocidas las causas del actual excepcional aumento de la supervivencia (¿mayor educación, hábitos saludables, nutrición, u otras?) en países desarrollados frente a los cazadores-recolectores actuales, que es superior a las 200 veces en ancianos (Vaupel, 2010; Burger et al, 2012).
La demencia en el anciano se achacó al envejecimiento, desde la civilización egipcia; y la medicina greco-romana se hizo eco de la misma, con alguna excepción como la de Cicerón (1924), con visión más benigna de la senectud, pues sugirió que una vida mental activa podría prevenir o posponer el declive cognitivo; visión que podemos firmar en la actualidad. Y hasta bien entrado el siglo XIX se hicieron muy pocos avances conceptuales en demencia porque ésta seguía considerándose una característica inevitable del envejecimiento (Berchold y Cotman, 1998). Y desde la perspectiva de la medicina naturalística, con Aristóteles a la cabeza, para la cual salud y enfermedad se distribuían de forma natural entre los seres vivos, es difícil de mantener que la demencia del anciano no sea sino una forma de envejecimiento exagerado del sistema nervioso. Sus egregios representantes actuales, ejemplo paradigmático, la “Bioestatistical theory” de Boorse y otros (Boorse, 2014; Kious, 2018), según la cual los fenómenos biológicos se distribuyen siguiendo parámetros de especie, edad y sexo con una distribución, digamos “gausiana”. Desde esa perspectiva, el declive cognitivo y la demencia del anciano sería una manifestación natural del envejecimiento, no una enfermedad (Brayne et al, 1995;Park et al, 2003; Singh-Manoux et al, 2010; Walhovd et al 2014) ¿Cómo considerar una enfermedad cuya incidencia crece de forma tan exponencial y análoga a la muerte en la naturaleza?, ¿Se puede considerar enfermedad una dolencia que afecta a un tercio de los mayores de 80 años, a un 25%-54% de los nonagenarios (Carrillo et al, 2008) y a quizá a más del 75% de los centenarios?, esto es la norma, no la excepción. (Brayne et al, 1995; Singh-Manoux et al, 2010; Breitner, 2014). Es cierto que las cifras de demencia y declive cognitivo aparente para los centenarios con buena salud son más benignas (45%), pero buena salud a esa edad no es la norma (Ailshire et al, 2024). Desde la perspectiva de la medicina naturalística no se podría sostener que la EA es una enfermedad.Pero estamos en tiempos y paradigmas de medicina normativa, y las normas médicas han establecido que sí lo es… Y, aun siguiendo esta ruta, muchos autores sostienen que declive cognitivo, demencia y EA están indisolublemente unidos al envejecimiento (Brayne, 1995, Lock, 2013; Ferrer, 2022), incluso a nivel básico, celular (Liu, 2022; Lau et al 2023). El estudio del envejecimiento y su relación con las enfermedades cerebrales como se lamentaba Adelman (1998), ha sido indebidamente postergado por la ciencia médica. Hay indicios de que en el futuro las cosas no van a ser así. Muchas entidades sociales están liderando el estudio del envejecimiento celular y molecular (Anti-Aging de Jeff Bezzos, Calico Lab, Hevolution Foundation, y otros) (Bermejo-Pareja y del Ser, 2024).
4. ¿Necesita la EA una redefinición?
Aunque esta consideración se aparta algo del tema central de este artículo, conviene exponer que el siglo XXI ha traído dos cambios conceptuales importantes en el ámbito de la demencia y EA.
1). Patología de la demencia y EA. Las series patológicas que actualmente recogen cerebros de los muy ancianos han mostrado, que su patología más frecuente son las causas mixtas: EA más lesiones vasculares, solas ambas, o asociadas a múltiples patologías tipo END (Schneider et al, 2007; Wharton et al 2023) entre las que destaca la LATE, acrónimo en inglés de “limbic-predominant age-related TDP-43 encephalopathy” (Nelson et al, 2019), establecida por innovaciones de las técnicas patológicas. La patología de la demencia del anciano (y EA) ha mostrado una gran multicausalidad patológica lo que dificulta que fármacos aislados tengan efectos terapéuticos en la demencia y EA del anciano (Jellinger, 2020; Nichols et al, 2023).
2). La disminución de la incidencia de demencia en países ricos occidentales (disminución no verificada en orientales como Japón o Corea del Sur) es una noticia muy positiva; el riesgo de demencia en las dos-tres últimas décadas ha disminuido debido (quizás) al efecto de la mayor educación de las poblaciones, mejora de las condiciones de vida desde la infancia, y terapia de los trastornos crónicos (Satizabal et al, 2016; Wu et al, 2017).
Estos dos hechos han propiciado: a) que la EA sea considerada más como un síndrome multicausal que como una enfermedad bien definida (Richard y Brayne, 2010). Zaven Khachaturian (2015), exdirector de estudios extramurales del NIA, y de la principal revista del campo de las demencias, lo sustenta; b) la prevención de la demencia del anciano se ha mostrado como una posibilidad plausible. Así, una comisión de la prestigiosa revista, “Lancet “, en varias publicaciones ha identificado 14 factores de riesgo, fundamentalmente clínicos, a los que se puede atribuir el 45% de los casos de demencia (Livingstone et al, 2017, 2020 y 2024). Otros estudios que analizan aspectos más amplios que los puramente clínicos, inciden en aspectos del desarrollo (maternos e infantiles), económicos y sociales a lo largo de la vida; y elevan esa cifra: 66% de contribución de factores ambientales en el desarrollo de demencia y EA (Xu et al, 2015; Bermejo-Pareja,2018; Weiss et al, 2020).
El estudio FINGER ensayo clínico multidimensional (Ngandu et al, 2015; Rosenberg et al, 2017) ha suscitado esperanzas porque los sujetos incluidos en la intervención preventiva retrasaban su declive cognitivo versus controles. En España, en Beasain (Euskadi) se ha realizado una experiencia piloto FINGER con resultados prometedores (Tainta et al, 2024). Estos estudios invitan a creer en la efectividad de intervenciones poblacionales para disminuir el riesgo de demencia como ha mostrado una revisión sistemática (Castro et al, 2023). Estos hechos animan a la implementación de políticas de salud pública locales y estatales y de los servicios de salud de cada comunidad (Bermejo-Pareja et al, 2016).
Lo que parece claro es que en el ámbito de la demencia del anciano se requiere de un nuevo paradigma científico en el que la investigación sobre el envejecimiento y la educación (reserva cognitiva) debieran ocupar un lugar central, así como las estrategias de prevención individual y poblacional. Es de desear que la investigación genética, epigenética y farmacológica faciliten el camino a un envejecimiento más satisfactorio con menor declive cognitivo, demencia y EA (Hachinski, 2023; Bermejo-Pareja y del Ser, 2024; Zhang et al, 2024).
5.). Conclusiones
La breve revisión de la terapia farmacológica de la EA pone de manifiesto la importancia de la hipótesis amiloide en el desarrollo de fármacos que eliminen el βA de del cerebro mediante inmunogénesis activa (vacunas), y posteriormente, mediante anticuerpos humanizados anti-βA. Ambas estrategias han fracasado tanto en la EA genética como en la esporádica. El lecanemab es un fármaco que no difiere de otros A-AβA ya utilizados, pero que ha suscitado gran controversia al ser aprobado por la FDA americana y no por la EMA. Juzgamos positiva la actitud de la EMA desde la perspectiva del paciente, de la ciencia y de la sociedad (coste elevado).
La discusión sobre el constructo nosológico actual de la EA concluye afirmando que la EA debe considerarse más como un síndrome multicausal muy ligado al envejecimiento que como una enfermedad sensu estricto.
Los datos recientes que indican una disminución del riesgo de demencia del anciano (y EA) apoyan la necesidad de una estrategia de prevención durante toda la trayectoria vital mediante una acción holística, médica y social y opuesta a la aprobación y acceso universal a tratamientos farmacológicos de dudosa eficacia y seguridad.
Referencias bibliográficas
Achenbaum WA. Robert N. Butler, MD (January 21, 1927-July 4, 2010): visionary leader. Gerontologist. 2014; 54:6-12. doi: 10.1093/geront/gnt015.
Ackley SF, Zimmerman SC, Brenowitz WD, et al. Effect of reductions in amyloid levels on cognitive change in randomized trials: instrumental variable meta-analysis. BMJ. 2021; 372:n156. doi: 10.1136/bmj.n156.
Adelman RC. The alzheimerization of aging: A brief update Exp Gerontol. 1998; 33: 155–157. doi: 10.1016/s0531-5565(97)00057-0.
Ailshire JA, Beltrán-Sánchez H, Crimmins EM. Becoming centenarians: disease and functioning trajectories of older US Adults as they survive to 100. J Gerontol A Biol Sci Med Sci. 2015; 70:193-201. doi: 10.1093/gerona/glu124.
Alzheimer A. Über eigenartige Krankheitsfälle des späteren Alters. Z Ges Neurol Psychiatr. 1911; 4:356-385. (Versión en inglés de Förstl H, Levy R. On certain peculiar diseases of old age. Hist Psychiatry. 1991; 2:71-101. doi: 10.1177/0957154X9100200505).
Assal F. History of dementia. Front Neurol Neurosci. 2019; 44:118-126. doi: 10.1159/000494959.
Bhadane P, Roul K, Belemkar S, Kumar D. Immunotherapeutic approaches for Alzheimer’s disease: Exploring active and passive vaccine progress. Brain Res. 2024;1840:149018. doi: 10.1016/j.brainres.2024.149018.
BallengerJF.; Self, Senility, and Alzheimer’s Disease in Modern America: A History;TheJohnHopkinsUniversityPress:Baltimore,MD,USA,2006.
Bartus RT, Dean RL III, Beer B and Lippa AS: The cholinergic hypothesis of geriatric memory dysfunction. Science. 1982; 217: 408‑417. doi: 10.1126/science.7046051.
Bermejo‐Pareja F. Alzheimer: Prevention from Childhood. Lamberth Academic Publishing; Mauritius. 2018.
Bermejo-Pareja F, Llamas-Velasco S, Villarejo-Galende A. Alzheimer’s disease prevention: A way forward. Rev Clin Esp (Barc). 2016; 216:495-503. (English & Spanish). doi: 10.1016/j.rce.2016.05.010.
Bermejo-Pareja F, Del Ser T. Controversial past, splendid present, unpredictable future: A brief review of Alzheimer disease history. J Clin Med. 2024; 13:536. doi: 10.3390/jcm13020536.
Boorse C. A second rebuttal on health. J Med Philos. 2014; 39: 683–724.doi:10.1093/jmp/jhu035.
Brayne C, Gill C, Paykel ES, Huppert F, O’Connor DW. Cognitive decline in an elderly population–a two wave study of change. Psychol Med. 1995; 25:673-83. PMID: 7480446.
Breitner JC. What should we do if we were wrong and Alzheimer was right? Int Psychogeriatr. 2014; 26:3-6. doi: 10.1017/S1041610213001749.
Burger O, Baudisch A, Vaupel JW. Human mortality improvement in evolutionary context. Proc Natl Acad Sci U S A. 2012; 109:18210-4. doi: 10.1073/pnas.1215627109.
Burke JF, Kerber KA, Langa KM, Albin RL, Kotagal V. Lecanemab: looking before we leap. Neurology. 2023; 101:661-665.doi:10.1212/WNL.0000000000207505
Butler RN. Age-ism: another form of bigotry. Gerontologist 1969; 9:243-6. doi: 10.1093/geront/9.4_part_1.243.
Carrillo-Alcalá ME, Bermejo-Pareja F. [Dementia in nonagenarians. Systematic review of population-based studies with Spanish data]. Rev Neurol 2008; 47:347-54 PMID: 18841545.
Castro CB, Costa LM, Dias CB, et al. Multi-Domain Interventions for Dementia Prevention – A Systematic Review. J Nutr Health Aging. 2023; 27:1271-1280. doi: 10.1007/s12603-023-2046-2.
CicerónMT. De senectute. Tricastela. Madrid. 2001 (Versión en latín y español).
Chowdhury S, Chowdhury NS. Novel anti-amyloid-beta (Aβ) monoclonal antibody lecanemab for Alzheimer’s disease: A systematic review. Int J Immunopathol Pharmacol. 2023; 37:3946320231209839. doi: 10.1177/03946320231209839.
Doody RS, Farlow M, Aisen PS; Alzheimer’s Disease Cooperative Study Data Analysis and Publication Committee. Phase 3 trials of solanezumab and bapineuzumab for Alzheimer’s disease. N Engl J Med. 2014; 370:1460. doi: 10.1056/NEJMc1402193.
Drachman DA, Leavitt J. Human memory and the cholinergic system. A relationship to aging? Arch Neurol. 1974; 30:113-21. doi: 10.1001/archneur.1974.00490320001001.
Ferrer I. Alzheimer’s disease is an inherent, natural part of human brain aging: an integrated perspective. Free Neuropathol. 2022; 3:3-17. doi: 10.17879/freeneuropathology-2022-3806.
Fox P. From senility to Alzheimer’s disease: the rise of the Alzheimer’s disease movement. Milbank Q. 1989; 67:58-102. PMID: 2682166.
Geylis V, Steinitz M. Immunotherapy of Alzheimer’s disease (AD): from murine models to anti-amyloid beta (Abeta) human monoclonal antibodies. Autoimmun Rev. 2006; 5:33-9. doi: 10.1016/j.autrev.2005.06.007.
Hachinski V; Dementia Prevention/Brain Health Initiative. We are preventing some dementias now-But how? The Potamkin lecture. Alzheimers Dement. 2023; 19:1067-1072. doi: 10.1002/alz.12770.
Hampel H, Mesulam MM, Cuello AC, et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain. 2018; 141:1917-1933. doi: 10.1093/brain/awy132.
Herrup K. How not to Study a Disease. The Story of Alzheimer’s disease. The MIT Press. Cambridge. Mass. 2021.
Høilund-Carlsen PF, Barrio JR, Werner TJ, Newberg A, Alavi A. Amyloid hypothesis: The Emperor’s new clothes? J Alzheimers Dis. 2020; 78:1363-1366. doi: 10.3233/JAD-200990.
Høilund-Carlsen PF, Alavi A, Barrio JR, et al. Donanemab, another anti-Alzheimer’s drug with risk and uncertain benefit. Ageing Res Rev. 2024; 99:102348. doi: 10.1016/j.arr.2024.102348.
Hoy SM. Lecanemab: First Approval. Drugs. 2023; 83:359-365. doi: 10.1007/s40265-023-01851-2.
Huang YM, Shen J, Zhao HL. Major clinical trials failed the amyloid hypothesis of Alzheimer’s disease. J Am Geriatr Soc. 2019; 67:841-844. doi: 10.1111/jgs.15830.
Itzhaki RF, Golde TE, Heneka MT, Readhead B. Do infections have a role in the pathogenesis of Alzheimer disease? Nat Rev Neurol. 2020; 16:193-197. doi: 10.1038/s41582-020-0323-9
Jellinger KA. Neuropathology of the Alzheimer’s continuum: an update. Free Neuropathol. 2020 Nov 11;1:1-32. doi: 10.17879/freeneuropathology-2020-3050.
Joseph J, Shukitt-Hale B, Denisova NA, Martin A, Perry G, Smith MA. Copernicus revisited: amyloid beta in Alzheimer’s disease. Neurobiol Aging. 2001; 22:131-46. doi: 10.1016/s0197-4580(00)00211-6
Katzman R. Editorial: The prevalence and malignancy of Alzheimer disease. A major killer. Arch Neurol. 1976; 33:217-8. doi: 10.1001/archneur.1976.00500040001001
Katzman R, Bick K. Alzheimer Disease. The Changing View. Academic Press. San Diego. 2000.
Kawas CH. Robert Katzman, MD. J Alzheimers Dis. 2009; 17:1-3. doi: 10.3233/JAD-2009-1039.
Khachaturian ZS, Khachaturian AS. Politics of science: progress toward prevention of the dementia–Alzheimer’s syndrome. Mol Aspect Med. 2015; 43‐44:3‐15. doi: 10.1016/j.mam.2015.06.001.
Kious BM.Boorse’stheory of disease: (why) do values matter? J Med Philos. 2018; 43: 421-438. doi:10.1093/jmp/jhy012
Kraepelin E. Psychiatrie: Ein Lehrbuch Fuer Studierende Und Aerzte. Verlag von Johann Ambrosius Barth. Lepzig.1910. Descrición en inglés de Schorer CE. Historical essay: Kraepelin’s description of Alzheimer’s disease. Int J Aging Hum Dev. 1985; 21:235-8. doi: 10.2190/gnq1-gdux-eptl-0f2l (el libro original en alemán se encuentra en la biblioteca del I. Cajal en Madrid).
Kuhn TS. The Structure of Scientific Revolutions. Second Edition. The University of Chicago. 1970.
Kurkinen M. Lecanemab (Leqembi) is not the right drug for patients with Alzheimer’s disease. Adv Clin Exp Med. 2023; 32:943-947. doi: 10.17219/acem/171379.
Kurkinen M, Fułek M, Fułek K, Beszłej JA, Kurpas D, Leszek J. The amyloid cascade hypothesis in Alzheimer’s disease: Should we change our thinking? Biomolecules. 2023; 13:453. doi: 10.3390/biom13030453.
Lanoiselée HM, Nicolas G, Wallon D, et al. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLoS Med. 2017; 14:e1002270. doi: 10.1371/journal.pmed.1002270.
Larson EB. Prevention of late-life dementia: No magic bullet. Ann Intern Med. 2018; 168:77-79. doi: 10.7326/M17-3026.
Lau V, Ramer L, Tremblay MÈ. An aging, pathology burden, and glial senescence build-up hypothesis for late onset Alzheimer’s disease. Nat Commun. 2023; 14:1670. doi: 10.1038/s41467-023-37304-3.
Livingston G, Sommerlad A, Orgeta V, et al Dementia prevention, intervention, and care. Lancet. 2017; 390:2673-2734. doi: 10.1016/S0140-6736(17)31363-6.
Livingston G, Huntley J, Sommerlad A, et al. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet 2020; 396:413-446. doi: 10.1016/S0140-6736(20)30367-6.
Livingston G, Huntley J, Liu KY, et al. Dementia prevention, intervention, and care: 2024 report of the Lancet standing Commission. Lancet. 2024; 404:572-628. doi: 10.1016/S0140-6736(24)01296-0.
LiuRM.Aging,cellularsenescence,andAlzheimer’sdisease. Int J Mol Sci. 2022; 23:1989.https://doi.org/10.3390/ijms23041989.
Lock, M. The Alzheimer Conundrum: Entanglements of Dementia and Aging; Princeton University Press: Princeton, NJ, USA, 2013.
Lozupone M, Dibello V, Sardone R et al. Lessons learned from the failure of solanezumab as a prospective treatment strategy for Alzheimer’s disease. Expert Opin Drug Discov. 2024; 19:639-647. doi: 10.1080/17460441.2024.2348142.
Mahase E. NICE rejects Alzheimer’s drug donanemab owing to cost and «significant health risks». BMJ. 2024; 387:q2342. doi: 10.1136/bmj.q2342.
Mullane K, Williams M. Alzheimer’s disease beyond amyloid: Can the repetitive failures of amyloid-targeted therapeutics inform future approaches to dementia drug discovery? Biochem Pharmacol. 2020; 177:113945. doi: 10.1016/j.bcp.2020.113945.
Nelson PT, Dickson DW, Trojanowski JQ, et al. Limbic-predominant age-related TDP-43
encephalopathy (LATE): consensus working group report. Brain 2019: 0; 1–25. doi: 10.1093/brain/awz099.
Ngandu T, Lehtisalo J, Solomon A, et al.. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): a randomized controlled trial. Lancet. 2015; 385: 2255–63.
Nicoll JAR, Buckland GR, Harrison CH, et al. Persistent neuropathological effects 14 years following amyloid-β immunization in Alzheimer’s disease. Brain. 2019; 142:2113-2126. doi: 10.1093/brain/awz142.
Orgogozo JM, Gilman S, Dartigues JF, et al. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology 2003; 61:46-54. doi: 10.1212/01.wnl.0000073623.84147.a8.
Pang M, Zhu L, Gabelle A, et al. Effect of reduction in brain amyloid levels on change in cognitive and functional decline in randomized clinical trials: An instrumental variable meta-analysis. Alzheimers Dement. 2023; 19:1292-1299. doi: 10.1002/alz.12768.
Park HL, O’Connell JE, Thomson RG. A systematic review of cognitive decline in the general elderly population. Int J Geriatr Psychiatry. 2003; 18:1121-34. doi: 10.1002/gps.1023.
Rapoport SI. Hypothesis: Alzheimer’s disease is a phylogenetic disease. Med Hypotheses 1989; 29:147-50. doi: 10.1016/0306-9877(89)90185-0
Richard E, den Brok MGHE, van Gool WA. Bayes analysis supports null hypothesis of anti-amyloid beta therapy in Alzheimer’s disease. Alzheimers Dement. 2021; 17:1051-1055. doi: 10.1002/alz.12379
Richards M, Brayne C. What do we mean by Alzheimer’s disease? BMJ 2010; 341:c4670. doi: 10.1136/bmj.c4670.
RitchieC.SmailagicN,Noel-StorrAH et al. PlasmaandcerebrospinalfluidamyloidbetaforthediagnosisofAlzheimer’sdiseasedementiaandotherdementiasinpeoplewithmildcognitiveimpairment(MCI). Cochrane Database Syst. Rev. 2014,CD008782.doi:10.1002/14651858.cd008782.pub4.
Robert C, Wilson CS, Lipton RB, Arreto CD. Evolution of the research literature and the scientific community of Alzheimer’s disease from 1983-2017: A 35-year survey. J Alzheimers Dis. 2020; 75:1105-1134. doi: 10.3233/JAD-191281.
RosenbergA,NganduT,RusanenM, et al.Multidomainlifestyleinterventionbenefitsalargeelderlypopulationatriskforcognitivedeclineanddementiaregardlessofbaselinecharacteristics:TheFINGERtrial. Alzheimer’s Dement. 2017;14:263–270.https://doi.org/10.1016/j.jalz.2017.09.006.
Satizabal CL, Beiser AS, Chouraki V, Chêne G, Dufouil C, Seshadri S. Incidence of dementia over three decades in the Framingham Heart Study. N Engl J Med. 2016; 374:523-32. doi: 10.1056/NEJMoa1504327.
Schiller ER, Silverglate BD, Grossberg GT. Profiling lecanemab as a treatment option for Alzheimer’s disease. Expert Rev Neurother. 2024; 24:433-441. doi: 10.1080/14737175.2024.2333970..
Schneider JA, Arvanitakis Z, Bang W, Bennett DA. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 2007; 69:2197-204. doi: 10.1212/01.wnl.0000271090.28148.24.
Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. 2016; 8:595-608. doi: 10.15252/emmm.201606210.
Sharma K. Cholinesterase inhibitors as Alzheimer’s therapeutics (Review). Mol Med Rep. 2019; 20:1479-1487. doi: 10.3892/mmr.2019.10374.
Singh-Manoux A, Kivimäki M. The importance of cognitive aging for understanding dementia. Age (Dordr). 2010; 32:509-12. doi: 10.1007/s11357-010-9147-7.
Smith AD. Anti-amyloid trials raise scientific and ethical questions. BMJ. 2021; 372:n805. doi: 10.1136/bmj.n805.
Steiner H, Capell A, Leimer U, Haass C. Genes and mechanisms involved in beta-amyloid generation and Alzheimer’s disease. Eur Arch Psychiatry Clin Neurosci. 1999; 249:266-70. doi: 10.1007/s004060050098.
Tainta M, Ecay-Torres M, de Arriba M, et al. GOIZ ZAINDU study: a FINGER-like multidomain lifestyle intervention feasibility randomized trial to prevent dementia in Southern Europe. Alzheimers Res Ther. 2024; 16:44. doi: 10.1186/s13195-024-01393-z.
Terry RD, Masliah E, Salmon DP, et al. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991; 30:572-80. doi: 10.1002/ana.410300410.
Terry N, Masliah A, Overk C, Masliah E. Remembering Robert D. Terry at a time of change in the world of Alzheimer’s disease. J Alzheimers Dis. 2019; 70:621-628. doi: 10.3233/JAD-190518.
Vaupel JW. Biodemography of human ageing. Nature. 2010; 464:536-42. doi: 10.1038/nature08984.
Van Dyck CH, Swanson CJ, Bateman RJ, et al. Lecanemab in Early Alzheimer’s Disease. N Engl J Med 2023; 388:9-21. doi:: 10.1056/NEJMoa2212948.
Weiss J, Puterman E, Prather AA, WareEB, Rehkopf DH. A data-driven prospective study of dementia among older adults in the United States. PLoS ONE. 2020 15: e0239994. doi : g/10.1371/journal.pone.0239994.
Wharton SB, Simpson JE, Ince PG, et al. Insights into the pathological basis of dementia from population-based neuropathology studies. Neuropathol Appl Neurobiol. 2023; 49:e12923. doi:10.1111/nan.12923.
Walhovd KB, Fjell AM, Espeseth T. Cognitive decline and brain pathology in aging–need for a dimensional, lifespan and systems vulnerability view. Scand J Psychol. 2014; 55:244-54. doi: 10.1111/sjop.12120.
Whitehouse PJ, George D. The Myth of Alzheimer’ Disease. What you aren’t being told about today’s most dreaded diagnosis. St Martin’s Griffin. New York. 2008.
Whitehouse PJ, Price DL, Clark AW, Coyle JT, DeLong MR. Alzheimer disease: evidence for selective loss of cholinergic neurons in the nucleus basalis. Ann Neurol. 1981; 10:122-6.
Wirak DO, Bayney R, Ramabhadran TV, et al. Deposits of amyloid beta protein in the central nervous system of transgenic mice. Science. 1991; 253:323-5. doi: 10.1126/science.1857970.
Wostyn P, Audenaert K, De Deyn PP. Alzheimer’s disease: cerebral glaucoma? Med Hypotheses 2010; 74:973-7. doi: 10.1016/j.mehy.2009.12.019.
Xu W, Tan L, Wang HF, et al. Meta-analysis of modifiable risk factors for Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2015; 86:1299-306. doi: 10.1136/jnnp-2015-310548.
Wu YT, Beiser AS, Breteler MMB, et al. The changing prevalence and incidence of dementia over time – current evidence. Nat Rev Neurol. 2017; 16:327-339. doi: 10.1038/nrneurol.2017.63.
Yankner BA, Lu T, Loerch P. The aging brain. Annu Rev Pathol. 2008; 3:41-66. doi: 10.1146/annurev.pathmechdis.2.010506.092044.
Zhang J, Zhang Y, Wang J, Xia Y, Zhang J, Chen L. Recent advances in Alzheimer’s disease: Mechanisms, clinical trials and new drug development strategies. Signal Transduct Target Ther. 2024; 9:211. doi: 10.1038/s41392-024-01911-3.
Joan Ramon Laporte, Mercedes Zurita, AVITE, Oriol Güell y NoGracias, reciben los Premios de la Asociación por un Acceso Justo al Medicamento. Manuel Rico (Investigate Europe), reconocimiento especial.
Palabras de Soledad Cabezón, presidenta de la AAJM: ”La actual política farmacéutica mata; atenta contra el Derecho Humano a la salud”.
Revista nº 34 Noviembre – Diciembre 2024
Comisión de Redacción de la rAJM
El catedrático de Farmacología Joan Ramon Laporte, la Dra. Mercedes Zurita (Hospital Universitario Puerta de Hierro), la Asociación de Víctimas de la Talidomida en España (AVITE), el periodista Oriol Güell (El País), la Organización Civil Internacional NoGracias y el periodista Manuel Rico, de la coalición europea Investigate Europe, recibieron el pasado 23 de noviembre los Premios de la Asociación por un Acceso Justo al Medicamento (AAJM).
El acto, que se celebró en el Ayuntamiento de Noblejas (Toledo), presidido por su alcalde, Agustín Jiménez Crespo, y la presidenta de la AAJM, Soledad Cabezón. Durante el mismo, se expresaron numerosas muestras de apoyo a las víctimas de la DANA, tanto de la comunidad autónoma de Valencia como de Castilla-La Mancha. Intervinieron también el Juan José Rodríguez Sendin, médico de Noblejas y ex presidente de la AAJM. El presidente de la Organización Médica Colegial. Tomás Cobo, envió un video de aliento a la labor de la AAJM.
Agustín Jiménez expresó su agradecimiento a la AAJM por volver a Noblejas como escenario de estos. En su intervención, hablo de enfermedad y pobreza, dos situaciones -afirmó- cuya alianza no es una casualidad porque “la enfermedad se ceba en la falta de recursos, en el hambre y en las carencias de un sistema sanitario insuficiente en recursos humanos y terapéuticos cuya pérdida progresiva de capacidad de respuesta -afirmó- no debemos seguir tolerando”.
“Por eso -añadió- apoyamos a la Asociación por un Acceso Justo al Medicamento porque pensamos que solo los ciudadanos bien informados y motivados serán capaces de cambiar una situación que origina la mayor morbimortalidad en el mundo junto con el mal reparto de la riqueza y la pobreza”. Aludió al crecimiento “desmesurado y sin control” del precio de los medicamentos que “ha agravado esta situación sin que los gobiernos, no solo no hagan nada para evitarlo, sino que sean cómplices de la misma”.
Premiados
Los Premios AAJM 2024, que este año celebran su II edición coincidiendo con el séptimo aniversario de la asociación, reconocen la labor de profesionales, instituciones, organizaciones y medios de comunicación que han destacado por su labor en defensa de la asistencia sanitaria de cobertura universal, del acceso justo a medicamentos y vacunas para todo el mundo y en la denuncia del uso abusivo de las leyes de propiedad intelectual que impiden el acceso a los medicamentos necesarios.
Joan Ramon Laporte, premio “Personalidad destacada en el ámbito político / sanitario” y Soledad Cabezón, presidenta de la AAJM. Acceso a vídeo PINCHANDO AQUÍ..
Joan Ramon Laporte, catedrático emérito de Farmacología de la Universidad Autónoma de Barcelona, recibió el premio “Personalidad destacada en el ámbito político/ sanitario” por su labor de denuncia entono a los ensayos clínicos y su crítica permanente y fundada a las prácticas de las empresas farmacéuticas.
Joan Ramon Laporte, tras agradecer el premio, resaltó la labor de la AAJM, “clave en el debate permanente sobre las políticas farmacéuticas”, y en especial la Revista de la AAJM que “ha contribuido de manera decisiva a su visibilidad”.
En su intervención habló de los riesgos por las fallas en los procesos de regulación y autorización de los medicamentos y dijo que “la mayoría de los nuevos medicamentos de precios exorbitantes no tienen pruebas convincentes de eficacia, y mucho menos de seguridad”. Asimismo, hizo referencia al sistema de patentes que “ha favorecido -dijo- la innovación comercial, pero no ha dado lugar a grandes avances en terapéutica” y se mostró partidario crear compañías farmacéuticas públicas, capaces de dar viabilidad al conocimiento generado desde el propio sistema sanitario.
Dra. Mercedes Zurita, premio “Mejor labor de una Institución científico-sanitaria” y Ángel María Martín Fernández-Gallardo, vicepresidente de la AAJM. Acceso a vídeo PINCHANDO AQUÍ. .
Dra. Mercedes Zurita, investigadora y responsable de la Unidad de Terapia Celular del Hospital Universitario Puerta de Hierro, recibió el premio a la “Mejor labor de una Institución científica-sanitaria”, por su trabajo de investigación de la terapia NC1 para pacientes con lesión medular, primer tratamiento de terapia avanzada y fabricación no industrial aprobado en España por la AEMPS que se ha convertido en uno de los referentes en investigación clínica.
Para Mercedes Zurita, este premio supone, “tanto para mí como para el resto del equipo de investigación que tengo el honor de dirigir, un reconocimiento a nuestro trabajo y a la dedicación que hemos tenido durante tantos años”.
La Dra. Zurita calificó las Terapias Avanzadas como la “medicina del futuro” y, tras destacar el gran potencial científico de la investigación en hospitales, cuestionó el hecho de que “muchos de desarrollos científicos que se hacen nuestros hospitales caigan en manos de farmacéuticas que al final son las que se llevan el reconocimiento y el dinero que generan las investigaciones realizadas al menos en parte con dinero público”. “Luego toca -añadió- comprar esas patentes a un alto precio para ponerlas a disposición de los pacientes por lo que el estado paga dos veces por lo mismo”.
Juan José Rodríguez Sendín, expresidente de la AAJM y Andrés Vizcaino que recogió el premio “Mejor labor de una Asociación de Pacientes” en nombre de la Asociación de Víctimas de la Talidomina de España (AVITE). Acceso a vídeo PINCHANDO AQUÍ. .
La Asociación de Víctimas de la Talidomida en España (AVITE) fue reconocida en la categoría de “Mejor labor de una Asociación de Pacientes”, por su lucha contra el imperio farmacéutico Grünenthal para conseguir el reconocimiento de las consecuencias que sufrieron a raíz del consumo de la talidomida en embarazadas, lo que provoco en los años 50 y 60 toda una tragedia con miles de casos de malformaciones congénitas y miles de muertes en todo el mundo.
Para Andrés Vizcaino, este premio a AVITE supone el “reconocimiento a una labor humanitaria en favor de un colectivo con severas discapacidades, ocasionadas por los efectos adversos de la talidomida”. Afirmo que “la evidencia de lo que provocó este medicamento ha servido para modificar legislaciones sobre autorización y comercialización de nuevos medicamentos, en favor de la protección de los pacientes, y los potenciales efectos secundarios”.
En su lucha constante para ayudar a las víctimas, Andrés Vizcaino, dijo que AVITE trabaja para que las ayudas no sean gravadas con un IRPF medio del 47%, lo que deja estas ayudas en la mitad y para que se aplique la Sentencia del Tribunal Supremo que ha dado la razón a AVITE, y reconoce otros 150 nuevos afectados, y que estos perciban las ayudas.
Fernando Lamata, expresidente de la AAJM y Oriol Güel premio “Mejor labor de información y divulgación”. Acceso a vídeo PINCHANDO AQUÍ.
El periodista Oriol Güell, responsable de temas de sanidad de El País, fue premiado en la categoría “Mejor labor de información y divulgación” por su labor informativa, rigurosa e investigadora en torno a sanidad pública, a la salud, y a la investigación, producción, comercialización y riesgos de diferentes medicamentos.
Oriol Güell agradeció a la AAJM por premiar su trabajo informativo como el de escribir sobre medicamentos y la industria farmacéutica que, “en ocasiones -afirmó- no es sencillo dada la complejidad de algunos temas y los fuertes intereses de los agentes implicados”.
Destacó la tendencia de los poderes públicos hacia una mayor transparencia sobre los precios de los medicamentos, cuya clave es “la voluntad política de todos los países y organismos internacionales implicados”. También hizo referencia al sistema de patentes y dijo que “a menudo las patentes no incentivan la innovación y el incentivo pasa conseguir extensiones poco justificadas de las patentes, a lo que se suma que la investigación proviene de centros púbicos”, por lo que “hay muchas razones que apuntan a que el sistema de patentes requeriría una revisión”.
Roberto Sánchez, que recogió en nombre de NoGracias el premio “Mejor labor de una Organización” y Roberto Sabrido, expresidente de la AAJM. Acceso a vídeo PINCHANDO AQUÍ.
Organización Civil Internacional NoGracias, recibió el premio a la “Mejor Labor de una Organización”, en la persona de su presidente Roberto Sánchez, por su labor crítica y de denuncia constante, a lo largo de más de 15 años, en defensa de la asistencia sanitaria universal y la transparencia en torno al buen gobierno de los medicamentos, así como la labor divulgativa para que los médicos no se dejen influir por los laboratorios.
Roberto Sánchez agradeció el premio a NoGracias por lo que supone un reconocimiento al trabajo de organizaciones que “representamos al interés público y que confrontamos con el gigante médico-industrial, a pesar de contar con pocos medios, problemas en el acceso a información clave y numerosas dificultades en el ejercicio de nuestra actividad”.
El presidente de NoGracias hizo referencia a la falta de transparencia en los precios de los medicamentos y tras preguntarse “¿Cómo puede ser que con nuestros impuestos se estén financiando bajo secreto comercial carísimos medicamentos? y ¿En qué otro sector de políticas es esto posible?”, afirmó que “el secreto es un instrumento que impone la industria para seguir manteniendo esos precios en la seguridad de que el escrutinio público sería incapaz de soportar y mantenerlos”.
Agustín Jiménez Crespo, alcalde de Noblejas y Manuel Rico, “Reconocimiento especial”. Acceso a vídeo PINCHANDO AQUÍ.
El periodista Manuel Rico, miembro de la coalición europea de periodistas de investigación Investigate Europe recibió un reconocimiento especial por toda su labor de investigación sobre el poder de las farmacéuticas y, en especial, por su trabajo para esclarecer lo ocurrido durante la primera ola de la COVID-19 en las residencias y apoyar a las familias de las víctimas.
Manuel Rico agradeció el reconocimiento al “trabajo colectivo que hemos realizado en Investigate Europe” y a la asociación que otorga este premio, que “trabaja para lograr justicia en un ámbito tan esencial como el acceso a los medicamentos para conseguir una sociedad más justa, algo por lo que estudié Periodismo, porque quería aportar mi granito de arena en ese camino”.
También se pronunció sobre la necesidad de poner fin a los elevados precios de los medicamentos y los beneficios desorbitados de la industria farmacéutica y dijo que para ello es preciso que haya “información, transparencia y mucha pedagogía” porque “la gente desconoce la realidad del mundo farmacéutico”. “Tener la voluntad de superar el problema, lograr suficiente influencia allí donde se toman las decisiones y llegar a otros ámbitos de la sociedad civil para evidenciar que son prácticas inaceptables”, es el camino, según Manuel Rico, para poner fin a esta situación.
Soledad Cabezón, presidenta de la AAJM.
Soledad Cabezón, presidenta de la AAJM, agradeció al Ayuntamiento de Noblejas, en especial a su alcalde, su “compromiso social manifiesto con la atención sanitaria a los ciudadanos” y su apoyo a la AAJM en “esta justa reivindicación para garantizar el derecho al acceso a la salud mediante el acceso a los medicamentos”.
Tras felicitar a los premiados, Soledad Cabezón expuso las características que definen la política farmacéutica actual (una intervención que de la que por el interés de su contenido recogemos a continuación un amplio resumen).
La actual política farmacéutica mata; atenta contra el Derecho Humano a la salud.
Según la OMS, un tercio de la población mundial carece de la posibilidad de obtener medicamentos esenciales (casi 2.000 millones de personas; mueren 10 millones de personas cada año, mayoritariamente en las poblaciones más pobres, y afecta más a personas de color, mujeres y niños).
En España y en la UE, una media de un 4% de la población, según Eurostat, declara no poder acceder a los tratamientos prescritos, donde el copago es un factor clave.
Es tal la problemática del acceso a los medicamentos que es hoy uno de los grandes retos de salud mundial. La Agenda 2030 lo recoge en su ODS nº3.
Los avances científicos y tecnológicos solo alcanzan a un 14% de la población del mundo, a EEUU, Japón y Europa, que consumen más del 80% de los medicamentos, mientras que el 86% restante de la población mundial sólo puede permitirse acceder al 20% de los medicamentos que se fabrican.
La OMS señala que un 93 % de la mortalidad evitable por enfermedades sucede en los países en vías de desarrollo, pero solo un 5 % de la investigación se dirige a problemas de salud de estas áreas por falta interés económico de la industria o falta de recursos (las denominadas enfermedades olvidadas, como son la tuberculosis o la malaria, erradicadas).
En cuanto a las enfermedades consideradas no transmisibles, causante principal de la mortalidad mundial, como la diabetes y las enfermedades cardiovasculares, la OMS señala también que solo alrededor de una quinta parte de las personas con hipertensión en el mundo reciben un tratamiento eficaz.
Podemos decir que la pandemia del VIH continúa más de 30 años después de que impulsase la Declaración de DOHA. Y es que, aunque se haya tratado a más de 22 millones de personas portadoras de VIH en el mundo, cada año casi un millón de personas sigue muriendo a causa del VIH/sida por falta de acceso a los tratamientos de última generación, como al lenacapavir.
Más cercano nos suena el que la OMS cifre en más de 40 millones de muertes innecesarias, o exceso de mortalidad, por falta de acceso a la vacuna contra el Covid19 durante la pandemia.
Por otro lado, la resistencia antimicrobiana es responsable de más 35000 muertes al año en la UE por falta de I+D en nuevos antibióticos. En 2050, de seguir la tendencia, un aumento continuo de la resistencia antimicrobiana podría dar lugar a 10 millones de muertes al año en todo el mundo, con una reducción del 2 al 3,5% del PIB mundial embargo, pero es que desde la década de los 90 del siglo pasado no se ha descubierto una nueva familia de antibióticos.
En definitiva, la industria investiga atendiendo a las reglas del mercado, siendo su fin el de los beneficios económicos y no los sociales, binomio incompatible con el derecho al acceso a la salud.
La política farmacéutica arruina nuestro sistema sanitario; amenaza su salud.
El negocio farmacéutico supone un 20% del gasto sanitario en la OCDE; según Lamata: «En España se gasta más en medicamentos que en todo la Atención Primaria. Entre noviembre de 2021 y 2022 aumentó un 5% el gasto farmacéutico. Eso son 616 millones de más en gasto farmaceútico ambulatorio que es lo que costaría contratar a 6.000 médicos «.
La OCDE ha proyectado que el crecimiento del gasto en salud de fuentes públicas será el doble del crecimiento promedio de los ingresos gubernamentales (2,6% y 1,3% respectivamente) entre 2019 y 2040. En consecuencia, el gasto en salud de fuentes públicas se prevé que alcance el 20,6% de los ingresos gubernamentales, un aumento de 4,7 puntos porcentuales con respecto a 2018.
Según IQVIA, a nivel mundial, en 2017 el gasto farmacéutico fue un 56% más que en 2007, con un crecimiento consecutivo del 12-14% cada quinquenio y se prevé que este será del 38% a 2028.
Este gasto se debe en gran parte a los altos precios de los nuevos medicamentos autorizados en las últimas décadas hasta llegar a ser inasequibles para muchos ciudadanos, incluso europeos, y amenazar seriamente la sostenibilidad de los sistemas sanitarios nacionales.
Los precios de los medicamentos no han dejado de incrementarse desde comienzos del siglo XX, pero especialmente a partir de finales de la segunda mitad con la aplicación de los derechos de propiedad intelectual creando monopolios de mercado, cuya generalización en el mundo se realiza a partir de 1995 con los Acuerdos de los ADIPC de la OMC.
Si el costo medio mensual del tratamiento contra el cáncer en 1960 era de 100 dólares en EEUU, pasó a más de 10.000 en 2012 y a más de 100.000 dólares por paciente y año para los nuevos tratamientos. Sin que, por otro lado, se haya acompañado de mejoras médicas proporcionales.
Precios, por otro lado, injustificados. Como recoge la publicación de Lamata y Gálvez, que indican que según las cifras publicadas por EFPIA (Federación Europea de Industria Farmacéuticas) sobre el gasto farmacéutico en la UE en 2016 de 179.535 millones de euros, si se hubiesen vendido a precio de genérico el coste habría sido un 41,5% de lo pagado, a lo que, si se le añadiese el gasto en I+D declarado por la misma fuente, EFPIA, de 26.913 millones, estiman que en la UE-28 hubo un exceso de gasto de 72.850 millones de euros sobre el precio de venta.
La política farmacéutica actual es sólo un modelo de negocio que adolece de ética.
Con, al menos, un 20% de retorno de inversión, el sector farmacéutico es uno de los sectores más lucrativos del mundo con el doble de margen beneficio en la UE que el resto de los sectores industriales.
Para muestra un botón; ya conocen ustedes como el recientemente comercializado inicialmente como tratamiento para la diabetes, pero utilizado masivamente para abordar la obesidad, ha colocado en riqueza a la empresa danesa Novonordisk por encima del PIB de su propio país Dinamarca en 2023. Pero en 2024, los ingresos netos suponen ya un 67% más que en 2023, con un 26% más de ganancias netas. Y lo que se espera…., a tenor de las noticias diarias sobre las múltiples posibles ampliaciones de indicaciones, incluido el alzheimer!, aunque adolezcan de los estudios adecuados, como sabe bien el catedrático en farmacología aquí presente, el Dr. Joan Ramón Laporte.
Mientras, el coste en general del desarrollo de estos y otros medicamentos se estiman en torno a 200 millones de dólares de media, según los datos facilitados por entidades sin ánimo de lucro, aunque la industria los cifra en 2870 millones de dólares, si bien se niegan a facilitarlos de forma transparente.
Datos más cuestionados aún para justificar los precios de los medicamentos si se tiene en cuenta la tendencia actual de adquisición por las industrias Big Pharma de lo que se ha venido a denominar empresas derivadas, de origen académico en su mayoría y financiación pública o por entidades sin ánimo de lucro, que, a la fecha, encabezan EEUU y el Reino Unido.
En el Reino Unido, la Universidad de Cambrigde ha desarrollado el 34% de ellas, mientras que la Universidad de Oxford el 13% de las empresas derivada, y cuyos costes de adquisición imputan como costes de investigación por parte de la industria.
Un ejemplo reciente y escandaloso lo tenemos en la vacuna contra el Covid de la empresa Moderna. El NIH (Instituto de Investigación Nacional de EEUU) acordó licenciar su tecnología a Moderna por 400 millones de dólares, lo que le revirtió a la empresa en 36 mil millones de dólares en ventas globales. Por su parte, el gobierno de EE.UU. invirtió al menos 31.900 millones de dólares para desarrollar, producir y comprar vacunas de ARNm contra el covid-19.
La política farmacéutica adoptada en el siglo XX es innecesaria para mejorar la salud.
Este modelo especulativo de la industria farmacéutica no fue siempre así, sino que es muy reciente, como el propio neoliberalismo.
El origen de la industrialización del producto farmacéutico se inicia a partir del siglo XIX, primero por parte de farmacéuticos y luego de personas procedentes de otra formación, como los químicos, que irán descubriendo nuevas sustancias que tendrán una aplicación terapéutica y a las que se les aplicarán procedimientos industriales con el desarrollo de la industria química; se produce así un tránsito progresivo de la fabricación artesanal de medicamentos en la rebotica de las farmacias a una elaboración industrial manufacturada del producto a escala. Durante el siglo XIX se van sucediendo descubrimientos de numeras sustancias químicas con aplicación terapéutica.
En América, un primer impulso de la industria farmacéutica se producirá tras la Guerra Civil (1861-1865), alcanzando un mayor grado de I+D, junto a Reino Unido, a raíz de la I Guerra Mundial (1914 -1918), debido a la interrupción del suministro de productos químicos alemanes y suizos. Aunque será la II Guerra Mundial (1939-1945) la que suponga el verdadero punto de inflexión hacia la industria farmacéutica moderna basada en I+D.
La II Guerra Mundial impulsa la fabricación a gran escala de la penicilina por las compañías farmacéuticas estadounidenses, cuya utilidad fue descubierta por el escocés Dr. Fleming en 1928, pero que hasta entonces se obtenía del cultivo de Penicillium en botes de cristal. El descubrimiento de la estreptomicina en el año 1944 por Marker supuso un enorme avance para el tratamiento de la tuberculosis con su desarrollo por el laboratorio Merck.
A este desarrollo industrial y surgimiento de numerosas moléculas se asiste sin la necesidad de derechos de propiedad industrial, aunque sí con un crucial apoyo público.
De nuevo, recordemos como en la historia reciente el apoyo público no ha sido menos crucial para impulsar la tecnología ARNm utilizada en las vacunas contra la Covid19, sobre la cual ya estaba realizada la investigación básica cuando irrumpe la pandemia; una I+D pública que servirá para el desarrollo de futuras y el impulso de nuevas terapias líneas de tratamiento. Pero también el desarrollo de su aplicación por parte del sector industrial contó con la inestimable financiación pública, cifrada en más de 5.800 millones de dólares, pero ahora ya bajo los derechos de propiedad intelectual, con miles de millones de beneficios para las empresas, pagados por los gobiernos (31.900 millones de dólares de factura en el caso de EEUU; aún estamos pendiente de los datos de la UE que la Comisión Europea se resiste a publicar) y que han dejado un exceso de mortalidad de 40 millones de personas en el mundo.
A finales de la primera mitad del siglo XX, en EEUU, la industria farmacéutica surgida en las reboticas, impulsada con el desarrollo de la industria química y posteriormente con el apoyo público durante las épocas de guerra, aunque no sólo, y que da lugar al surgimiento de empresas como Lilly o el auge de Novartis, comienza a presionar para que se apliquen los derechos de propiedad intelectual al medicamento.
A partir de finales de la década de los años sesenta del siglo XX, comienza a apreciarse una fuerte tendencia de los países desarrollados, en general, por incorporar los medicamentos en los sistemas de patentabilidad para proteger a sus industrias. En los países en vía de desarrollo será más tarde, a finales de la década de los 1980, con las negociaciones de la Ronda Uruguay.
En Europa, los derechos de propiedad intelectual se empiezan a incorporar al medicamento más tarde; en 1967 en Alemania, en 1968 en Francia, 1977 en Suiza y 1980 en Italia.
En España, la Ley de Propiedad Industrial de 1929 lo prohibía expresamente y no será hasta 1986, tras la firma del Tratado de Adhesión a la Comunidad Europea, cuando se introduzca la patente del medicamento en nuestro país.
Sin embargo, en 1987, la Secretaría de la OMPI preparó un informe sobre los sectores tecnológicos excluidos de la patentabilidad en las diferentes legislaciones nacionales, siendo habitual encontrar al farmacéutico entre los 19 sectores habitualmente excluidos. Este informe servirá de base para lo que vino después, los Acuerdos de los ADIPC, adoptados en 1995 en la OMC, en el contexto de la globalización económica, que vendrán a establecer a nivel internacional normas comunes en torno al comercio y extenderán y ampliarán la aplicación de forma obligatoria de los derechos de propiedad intelectual a todas las áreas del conocimiento, incluyendo, ahora sí, a los medicamentos.
La patente del producto farmacéutico supondrá un monopolio para la industria farmacéutica, con exclusividad de mercado durante años, eliminando la competencia, con el fin de recuperar la inversión económica realizada en el desarrollo del producto y estimular así la I+D. Queda claro que este sistema ha contribuido claramente en que el sector farmacéutico se haya convertido a día de hoy en el más competitivo de la UE, como también el que la factura farmacéutica ha ido creciendo hasta representar entorno al 20% del gasto en salud en los países de la UE y la OCDE y una media 1,5 % respecto del PIB.
Sin embargo, no parece que haya estimulado la I+D en cantidad ni en calidad. Un estudio llevado a cabo por la Universidad de Harvard en 2009 señalaba que el número de moléculas nuevas no conocidas no sólo era mayor previamente a los Acuerdos de los ADIPC, sino que tras su aprobación el número de nuevas sustancias fue menor e incluso, viendo la evolución de la serie, pareciera como si se hubiese desincentivado la calidad innovativa, no recuperándose nunca el número de nuevas sustancias de la década de los 70.
La OCDE también publica otra serie en la que la media de autorizaciones por la FDA de nuevos productos, concepto más amplio que el de nuevas sustancias, se ha mantenido estable desde 1980 a 2014, en torno a 100 al año.
En 2015, la consultora IQVIA estimaba en una media de 46 aprobaciones de nuevos medicamentos al año por la FDA en la última década y así lo preveía hasta 2028. Del mismo modo, el Institute for Healthcare Informatics (IMS) no observa variaciones significativas desde 2005 a 2018 en la aprobación de nuevas sustancias. En cuanto a las cifras en la UE publicadas, los nuevos medicamentos se aproximarían a los 40. Sin embargo, si se limitase a las sustancias realmente nuevas, sería la mitad.
Sin embargo, lo llamativo es como el número de nuevas sustancias dista mucho con el número de patentes de productos farmacéuticos concedidas en estos años por la UPSTO y la OEP, especialmente tras la entrada en vigor de los ADIPC.
Según los datos de la base de datos de la OCDE, la aprobación de los Acuerdos de los ADIPC supuso un llamativo incremento del número de aplicaciones de modificaciones o variaciones de productos previamente autorizados, en total se han registrado 77.645 aplicaciones de variaciones tipo I y 20.853 de tipo II, y de patentes en general, contabilizadas en miles desde 1995, y no de nuevas sustancias. Es decir, este acuerdo vino a dar lugar a un modelo de negocio muy lucrativo, pero nada innovador y, por lo tanto, innecesario e injustificado, a diferencia del mantra mantenido por los defensores del modelo vigente y la necesidad de los derechos de propiedad intelectual para estimular la I+D farmacéutica.
En definitiva, mientras que el número de patentes de medicamentos se cuentan en miles, se evidencia una falta de innovación genuina. Detrás de este alto número de patentes se encuentra la estrategia denominada “maraña de patentes” como modelo de negocio que mediante pequeñas modificaciones de los productos para alargar la vida de la patente se perpetúan las ganancias impidiendo la entrada de los genéricos al mercado. La Comisión Europea en su informe de 2008, único en el que estudió y publico él funcionamiento del mercado farmacéutico de forma global en la UE pero que sigue tan vigente como entonces, señaló que se daba el caso incluso de que un medicamento obtuviese 1300 patentes y cifraba que más del 78% de las patentes no se trataban de nuevos medicamentos, infravalorado este porcentaje si se atendiese al número de nuevas moléculas o principios activos.
En 2022, I-MAK señalaba que los 10 medicamentos más vendidos en EEUU habían obtenido una media de 74 patentes. Por otro lado, la compañía AbbVie, comercializadora de Humira, el medicamento más caro en 2022, de las 312 solicitudes de patentes sobre el medicamento que se presentaron en la FDA, obtuvo 166.
Además, un estudio sobre los medicamentos que fueron introducidos en el mercado francés entre los años 2006 y 2011, llegó a la conclusión de que el número de moléculas que aportaron un progreso terapéutico importante, disminuyó drásticamente de 22, en el año 2006 a 15, 10, 7, 4 en los años siguientes.
El futuro farmacéutico es ya el presente y no parece alentador. Se resiste una I+D pública.
Detrás de todo este entramado de negocio bursátil se da una problemática que atenta contra el derecho a la salud al impedir el acceso a los medicamentos. Y lo que se consideraba un problema de los países en desarrollo, que ya se opusieron a los Acuerdos de los ADIPC de 1995 por las consecuencias en los precios que acarrearía y que ya Correa anunciaba entonces, ha llegado a ser efecto común a todo el sistema, afectando también a los países desarrollados; sólo era cuestión de tiempo ante un sector que maximiza ganancias como única estrategia.
En 2014, el mundo desarrollado europeo toma conciencia real de la problemática sobre el acceso a los medicamentos al no poder garantizar el acceso al sofosbuvir, medicamento que permitía erradicar la hepatitis C, debido a su alto precio. Ello determina un punto de inflexión que abre definitivamente el, hasta entonces soterrado, debate sobre la necesaria revisión del funcionamiento del sistema farmacéutico europeo.
El Parlamento Europeo toma la iniciativa mediante la elaboración del primer informe que revisa a nivel europeo la problemática del AM en la UE, señala sus disfunciones y eleva propuestas de mejora.
Al mismo tiempo, el Consejo de Ministros de Salud de la UE celebrado en Ámsterdam en julio de 2016, solicita por primera vez un requilibrio del sistema farmacéutico de la UE, haciendo especial hincapié en la necesidad de revisar el impacto de la propiedad intelectual y en la implementación e impacto de los incentivos a la I+D.
Hasta ahora, en la UE, el mercado farmacéutico se ha caracterizado por un alto nivel de regulación para garantizar su libre circulación como mercancía, pero desde el ámbito de la salud, más allá de la seguridad, no ha recibido la misma atención la calidad de la innovación y, menos aún, el aspecto social de garantía de acceso, en términos de disponibilidad, asequibilidad y accesibilidad, dejando al sector empresarial la capacidad de decidir sobre la priorización de las líneas de investigación en base al mercado y estableciendo su precio.
La amenaza de la pandemia contra el Covid19 supuso un impulso en el objetivo de la UE en salvaguardar la salud pública y vino a definir la Unión Europea de la Salud, que englobó el Pilar Social Europeo. El acceso a los medicamentos fue recogido entonces, por primera vez por parte de las instituciones europeas, como uno de los retos en salud y al que se debe dar respuesta, lo que pretende llevar a cabo dentro de la Estrategia Farmacéutica Europea publicada en 2022 por la CE.
Esta Estrategia Farmacéutica Europea vendrá a determinar el funcionamiento del sistema farmacéutico en las próximas décadas; su acierto, o desacierto pueden tener consecuencias económicas, pero también y, especialmente, en términos de salud y bienestar para la ciudadanía europea.
Por otro lado, el siglo XXI es el siglo de la «revolución tecnológica y digital». La salud tampoco puede ni quedará al margen de los desafíos y oportunidades de esta nueva era del desarrollo científico y tecnológico. Sin embargo, este paradigma puede ser integrado como parte de la respuesta a los retos a los que se enfrenta la sanidad o ahondar en un modelo de negocio que genere nuevos obstáculos y dé origen a nuevas desigualdades.
El desarrollo de terapias avanzadas desde el sector público, como se reconoce hoy aquí en la figura de la Dra. Zurita, representa un ejemplo para que los EEMM, las instituciones europeas, los gobiernos nacionales y la sociedad civil se conciencien del papel y oportunidad que, en este contexto de revisión del modelo farmacéutico, brinda una farmacia pública en una nueva era de la medicina, denominada de precisión, para garantizar la sostenibilidad de los SNS, la equidad y cohesión social.
Esta revisión integral de la legislación farmacéutica europea abre una oportunidad única para la reflexión sobre el funcionamiento del modelo de I+D farmacéutico y su reformulación, en su caso. Sin embargo, hasta ahora, las propuestas presentadas por la CE han ido encaminadas hacia una perpetuación e incluso fortalecimiento del sistema vigente, en lo que el Parlamento Europeo ha ahonda aún más.
Especial mención quiero hacer sobre el Espacio Europeo de Datos Sanitarios, incluido dentro del paquete legislativo de la Estrategia Farmacéutica Europea, cuya formulación supone una descapitalización de los sistemas sanitarios públicos con la transferencia altruista al sector privado de la información recopilada a lo largo de décadas y que aumentará de forma exponencial con el desarrollo tecnológico, cuyos resultados derivados la I+D realizada con los mismos los volverá a pagar el propio sistema sanitario público mediante la adquisición de las terapias.
Por qué no desarrollar decididamente un sistema público de investigación que normalice lo llevado a cabo por la Dra. Zurita en el Hospital Puerta de Hierro o Joan Manel en el Hospital Clinic de Barcelona con las CAR-T. De momento, en España, no hemos más que asistido a la puesta en marcha de una iniciativa de empresa público-privada dentro del PERTE de Salud, cuyas condiciones de explotación y beneficios no quedan claras en lo que respecta al sector público.
Desde la AAJM abogamos por una investigación farmacéutica pública y para ellos nos basamos en datos como los publicados por la OCDE sobre 2014 y 2018 que señalan que la financiación pública de la I+D farmacéutica es superior al 40% en la UE; lo que representó en 2014 un 43% de financiación pública respecto de la privada, un 0,14% y un 0,06% del PIB respectivamente, y del 67,5% en 2019, lo que supuso un 0,1% y un 0,07% del PIB respectivamente.
Sin embargo, todo apunta a que no sólo se incentivará una I+D pública para dar respuestas a las necesidades médicas no cubiertas al rechazarse de forma explítica la iniciativa en forma de enmienda firmada por 50 eurodiputados en el Parlamento Europeo, sino que el objetivo principal no será mejorar el acceso a los medicamentos, sino dar respuesta a una supuesta pérdida de competitividad del sector industrial farmacéutico ante la emergencia de países competidores de la región asiática.
Es cierto que hace 25 años 1 de cada dos medicamentos nuevos se originaba en Europa, mientras que ahora es 1 de cada 4 o 5 y que en 2022, en China y en la EU se originaron 18 y 19 nuevos medicamentos, respectivamente. Sin embargo, nada se dice que China, uno de los competidores principales que se asoman al mercado internacional de medicamento, invierte en I+D farmacéutica una décima parte de lo que declara EEUU y una quinta parte que la UE, lo que apuntarían a un sistema más eficiente, a falta de conocer lo que se imputa en ID otras actividades en EEUU y la UE, al mismo tiempo que pareciese que lo que realmente se pretende es mantener un alto nivel de retorno de la inversión, un sector altamente lucrativo.
España
No quiero dejar de referirme a España, pues en nuestro país, el Gobierno, ha abierto revisiones o impulsado normativas respecto de la política farmacéutica que pueden suponer una oportunidad para mejorar el AM y la sostenibilidad de los SNS.
La revisión de la Ley de Garantías y de Uso Racional del Medicamento puede ser una oportunidad para introducir una experiencia piloto de I+D pública, así como para mejorar el uso de medicamentos genéricos y ahorro del gasto farmacéutico eliminando el margen de beneficio destinado a la farmacia, por ejemplo.
La Ley de Equidad para eliminar los copagos y derogar definitivamente los coletazos pendientes del “RD 2012”.
El Reglamento Sobre Terapias Avanzadas (SOHO), recientemente revisado en la UE bajo la presidencia española del Consejo, para la introducción en él de las CAR-T y protegerlas del riesgo de limitar la exención hospitalaria con la que amenaza la Estrategia Farmacéutica Europea Europea y que, además, limitaría el desarrollo público de la ID.
El Real Decreto Sobre Evaluación de Tecnología Sanitaria que propone la transparencia en los costes de los costes de I+D y desarrollo global de los medicamentos pero que debería concretarse hacia su uso en el establecimiento de precios.
Foto de familia de los participantes en el acto de los premios AAJM 2024.
A modo de conclusión
Juan José Rodríguez Sendín, médico de Noblejas durante 36 años, ex presidente de la OMC y uno de los impulsores de la AAJM, puso en valor estos premios con los “queremos impulsar -dijo- la sensibilidad ciudadana en la defensa del medicamento como bien social, un derecho humano preferente al que deben tener acceso todas las personas en condiciones justas” y agradeció al alcalde de Noblejas su implicación en esta causa y en la defensa de la atención sanitaria.
Rodríguez Sendín habló del “incremento de la inequidad social que ocurre -dijo- al limitar la capacidad del sistema sanitario para mejorar la salud”. Y, tras cuestionar los “tratamientos inútiles y tecnologías en casos y en pacientes en los que no están indicadas” que está soportando el Sistema Nacional de Salud por “comportamientos de los Gobiernos sanitarios e intereses contrarios al interés del paciente”, abogó por el deber del médico para “ajustar sus actos y decisiones exclusivamente a la necesidad sanitaria del paciente”.
Conferencia impartida el 13 de noviembre de 2024 en las jornadas organizadas por el Comité de Ética de la Investigación del Instituto de Salud Carlos III, X Jornadas sobre Aspectos Éticos de la Investigación Biomédica: “Repensar las cuestiones éticas para una investigación global”.
OTRAS FUENTES. Revista nº 34 Noviembre – Diciembre 2024
En esta conferencia abordaré algunos aspectos éticos que tienen que ver con la financiación de la investigación por el Sistema Nacional de Salud (SNS).
1. Financiación de la investigación biomédica en el SNS.
El Sistema Nacional de Salud financia la I+D de dos maneras. Una es la financiación pública directa. La otra es a través de las patentes y otras exclusividades.
1.1.
La financiación pública directa de I+D biomédica en España (la que hacemos con dinero del presupuesto público) ascendió a 3.543 M€ en 2021, según el Informe sobre el sistema sanitario del Consejo Económico y Social, publicado en marzo pasado (1).
El Instituto de Salud Carlos III, organizador de estas Jornadas, es el principal organismo encargado de la gestión y ejecución de los programas de I+D biomédica. Hay, además, 35 Institutos de Investigación Sanitaria, formados por hospitales y centros de salud, universidades y otras instituciones, que reúnen 29.000 investigadores adscritos. Además, hemos de sumar los organismos de investigación de las CCAA. El ISCIII gestiona los Centros Nacionales de Investigación, los Centros Propios, las redes de investigación cooperativa orientadas a resultados en salud, la Plataforma de Soporte para la Investigación Clínica, y el seguimiento y apoyo a los centros de Investigación Biomédica en Red, los CIBER.
En España, la financiación pública directa de investigación biomédica asciende a dos tercios del total. Por su parte, en la UE, dicha financiación asciende al 45% (2). Y en EEUU, con los Institutos Nacionales de Salud, a más de un tercio del total (3).
Como ha señalado la profesora Mariana Mazzucato, la mayor parte de la investigación básica y buena parte de la investigación clínica innovadora se realiza en centros públicos o con financiación pública (4). La investigación con financiación pública directa es determinante. Sin embargo, el control y la explotación de los resultados del sistema de investigación se realiza, principalmente, por la industria farmacéutica. Empezando por controlar la narrativa: pareciera que toda la investigación biomédica esté financiada por las empresas farmacéuticas. Y, como se ha visto, no es así.
1.2. La financiación de la industria.
Según la memoria de Farmaindustria, en el año 2022 las compañías farmacéuticas destinaron 1.395 millones de euros a investigación biomédica en España (5). Un tercio de la I+D biomédica total.
Con estos fondos las empresas pagan ensayos clínicos, investigación básica y otros gastos relacionados. Ahora bien, y esto es muy importante, esta parte de la financiación de la I+D, la financiación pública indirecta, que aparentemente pagan las compañías farmacéuticas, también la financiamos los contribuyentes y los pacientes, con los sobre-precios que exigen las empresas por los nuevos medicamentos, gracias a los monopolios que conceden los gobiernos, a través de las patentes y otras exclusividades. Por eso la defino como financiación pública indirecta.
1.3. El modelo de patentes y monopolios para financiar la I+D.
Analicemos brevemente este mecanismo, que es clave. El monopolio que permiten las patentes es una especie de impuesto cedido a las empresas. Por lo tanto, es dinero público que no figura en el presupuesto y que, como enseguida veremos, genera una serie de conflictos éticos y efectos secundarios que pueden ser negativos para el Sistema nacional de salud.
Las patentes y otras exclusividades se otorgan por los gobiernos a las empresas farmacéuticas, prohibiendo durante un determinado número de años la comercialización de productos genéricos y biosimilares por los competidores, de manera que las empresas originarias puedan poner precios más altos, sin competencia.
La justificación de que los gobiernos concedan estos monopolios es la financiación de la I+D. Como dije antes, actuarían como una especie de impuesto indirecto que los gobiernos permiten fijar y cobrar a las compañías farmacéuticas para financiar, supuestamente, la investigación. Por eso les llaman “incentivos” para las empresas.
El problema es que, sistemáticamente, se ha producido un abuso de posición dominante, un abuso del monopolio, exigiendo las empresas precios mucho más altos de lo que sería necesario para financiar la I+D que han realizado. De esta forma, obtienen unos beneficios exagerados, parte de los cuales destinan a marketing y acciones de lobby, para mantener su posición. De hecho, gastan más en marketing que en I+D. Otra parte la destinan a recompra de acciones y otras fórmulas para remunerar a los accionistas y los ejecutivos. También destinan a estos cometidos más dinero que a I+D. Y los gobiernos lo permiten, aunque esa no fuera la finalidad del “incentivo”, que se ha pervertido progresivamente.
Siguiendo la metodología del profesor Dean Baker (6), para estimar qué cantidad suponen los sobre-precios, calculamos lo que pagaríamos por todos los medicamentos a precio de genérico o biosimilar. Así, en España, el sobre-precio en medicamentos que pagó el SNS y pagamos los pacientes, por encima de los costes de fabricación y de un beneficio industrial medio, ascendió a 11.792 M€ en 2022, y de esa cantidad, como hemos visto, las empresas solo destinaron a I+D 1.395 M€. El resto, 10.397 M€, se lo embolsaron como beneficios, que destinaron a otros fines.
Es decir, en España, el “incentivo” a la investigación que abonamos los contribuyentes y los pacientes, no solo paga el 100% de todo lo que dicen que se gastaron las empresas en I+D. Sino que paga siete veces más (7, 8, 9, 10).
¿Por qué las empresas suben los precios de los nuevos medicamentos exageradamente con el monopolio? Porque quieren maximizar sus ganancias, y porque los pacientes necesitan ese producto. Les va la vida en ello. Y una madre da todo lo que tenga si es para salvar a su hijo. En otros productos, en otros bienes y servicios, en los que las empresas disfrutan de un monopolio durante un tiempo, un nuevo teléfono móvil, un nuevo coche, una nueva máquina, los consumidores podemos tomar la decisión de prescindir de ese producto y consumir el antiguo hasta que la empresa baje el precio o pierda la patente y surja un competidor. En cambio, cuando se trata de un medicamento, si es un paciente privado, se endeudará hasta declararse en quiebra, y, si es un sistema de salud, los pacientes le presionarán para que el gobierno acepte pagar lo que le pidan.
Con esa premisa, y con el monopolio, los directivos de las empresas tenderán inevitablemente a poner el precio más alto posible, lo más alto que puedan pagar los consumidores. Porque se lo exige la lógica del mercado, la codicia. Se lo exigen sus accionistas principales que, en las grandes empresas farmacéuticas, son enormes gestoras de fondos de inversión, como Blackrock Finance, State Street Corporation, Vanguard group, y otras entidades financieras. El objetivo declarado de estos grandes accionistas es maximizar beneficios.
El medicamento se convierte así en un producto financiero más. Además de las ganancias por ventas, se genera otro enorme beneficio por revalorización de acciones, que en no pocas ocasiones se produce por la recompra que hacen las mismas empresas de dichas acciones con las ganancias abusivas de los sobre precios.
Es decir, el mecanismo de financiación de la I+D de nuevos medicamentos a través de monopolios (patentes y otras exclusividades), lleva inevitablemente al abuso de posición dominante.
Las empresas dicen que las patentes son la sangre de la innovación. Ya hemos visto que la mayor parte de la innovación proviene de I+D con financiación pública directa. Pero tampoco es cierto que sin patentes las empresas no investigaran. Conviene recordar aquí que antes de la generalización de las patentes de medicamentos, que se produjo con el acuerdo sobre los Aspectos de los Derechos de la Propiedad Intelectual relacionados con el Comercio, el ADPIC, en 1994, las empresas farmacéuticas investigaban. Y ganaban dinero. De hecho, entre 1950 y 1995, el promedio de beneficios sobre ventas fue del 10%, el doble de lo que obtuvieron la media de las industrias no farmacéuticas. Lo que ocurrió a partir de 1995, es que los beneficios sobre ventas se dispararon, al 20, al 30, al 50 por ciento sobre ventas, o más. Pero las empresas no aumentaron su investigación innovadora, sino que priorizaron sus objetivos comerciales y la compra de resultados de investigación hecha en centros públicos.
Los lobbies de la industria insisten en que los nuevos medicamentos son “de alto coste” porque la investigación cuesta mucho. Es cierto que cuesta mucho. Pero los precios de esos nuevos medicamentos están muy por encima de los costes de fabricación y de I+D, como ya vimos para España. En realidad, son medicamentos de “alto precio”, más que de “alto coste”.
Por ejemplo, como ha demostrado Andrew Hill, profesor de la Universidad de Liverpool, los antivirales de acción directa para la Hepatitis C tienen un coste de fabricación y de I+D de menos de 300 euros por tratamiento, y en España hemos pagado un precio de 20.000 euros por tratamiento. Así ocurre con otros muchos medicamentos (11, 12, 13, 14). Por culpa de los monopolios, se están aprobando precios de más de 10.000 ó 100.000 euros por tratamientos cuyo coste, incluida la I+D es cinco, diez o cien veces menor.
Merece la pena recordar que, en septiembre de 2020, la señora Carolyne Maloney presidía la sesión del Comité de Vigilancia y Reforma del Congreso de los Estados Unidos, donde se llevó a cabo una investigación sobre los altos precios de los medicamentos. Citó el caso de Anoinette Worsham, que había perdido a su hija diabética, al tener que racionar la insulina que necesitaba, porque no podía pagarla. En aquella sesión la congresista Maloney afirmó: “Los documentos revisados por el Comité (más de un millón de páginas) muestran que estos aumentos masivos de precios se basan en generar enormes beneficios para estas compañías, sus accionistas y sus ejecutivos -y añade-. Hemos oído los argumentos de las compañías farmacéuticas y sus lobistas defendiendo la necesidad de subir precios para pagar la investigación en medicinas que salvan vidas… pero la investigación del Comité muestra que esos argumentos son completamente falsos”.
Tengamos también en cuenta que en el gasto de I+D que dicen que han realizado las industrias está toda la investigación, la que funciona y la que no (además de gastos en empresas intermediarias de gestión de ensayos clínicos, ensayos clínicos promocionales, ensayos en medicamentos me-too, bufetes de abogados especializados en patentes, etc., etc.).
2. Problemas éticos que provoca la financiación de los medicamentos en el SNS.
Una vez analizado el modelo de financiación de la investigación en medicamentos a través de patentes, veamos ahora cómo influye este modelo en seis importantes aspectos del Sistema Nacional de Salud.
2.1.
El primer aspecto que podemos comentar, con claras implicaciones éticas, se refiere a la asignación de recursos. Como hemos visto, el modelo de financiación de la I+D de nuevos medicamentos, a través de monopolios y sobreprecios, provoca gastos excesivos e innecesarios a los Sistemas de Salud, implicando un riesgo para su viabilidad, y afectando al derecho a la atención sanitaria y a la salud de las personas.
Porque el dinero público es limitado. Y lo que gastemos de más en medicamentos no se puede destinar a otros fines muy necesarios. En 2022 el gasto sanitario público total ascendió a 92.072 M€. De esta cantidad, 22.015 M€ se gastaron en medicamentos. Es decir, el gasto en medicamentos supuso un 24% sobre el gasto sanitario público total, cuando debería ser como máximo la mitad. Y sigue creciendo.
Mientras aumentaba el gasto farmacéutico, la sanidad pública en España se iba deteriorando. Los tiempos de espera para recibir atención, o para ser operado, se han duplicado en los últimos diez años. Por otro lado, la valoración de la población sobre la sanidad pública ha caído de un 74% que consideraban que funcionaba bien o muy bien, a un 55,8%. Y, al mismo tiempo, el número de personas que utiliza una póliza sanitaria privada se ha duplicado, de un 16% a un 35% de la población.
Ese exceso de gasto farmacéutico, de más de 10.000 M€ anuales, que se desvía a los accionistas de la industria farmacéutica podría servir para mejorar radicalmente la Salud Pública, la Atención Primaria, la atención en Salud Mental, las condiciones de trabajo de los profesionales, los tiempos de espera y de atención, así como para consolidar los centros públicos de investigación y generar alternativas públicas para la fabricación y desarrollo de medicamentos innovadores y medicamentos esenciales.
Para dimensionar el exceso de gasto por abuso de patentes, podemos decir que equivale a poder contratar a más de 100.000 profesionales sanitarios; o que es equivalente al funcionamiento de 10.000 camas de hospital, o de 1.700 centros de salud dotados de personal y equipamiento.
Además, con ese dinero se podría reforzar el sistema público de investigación, potenciando la investigación en estrategias de prevención y promoción de la salud. No olvidemos que la prevalencia ajustada por edad del cáncer, o de los trastornos mentales y otros procesos, ha crecido más de un 30% en los últimos diez años.
A ese exceso de gasto por sobre precios se puede añadir el gasto por sobre prescripción, que luego comentaré, estimado en unos 5.000 M€ anuales para el conjunto de la sanidad.
2.2.
El segundo problema ético, derivado de los medicamentos de altos precios, es el problema de acceso.
En efecto, en España, con un buen sistema de salud, y un nivel de renta de país desarrollado, tenemos problemas de acceso a medicamentos.
En primer lugar, por los copagos.
Según el último barómetro sanitario, en los últimos 12 meses, un 4% de la población dejó de tomar el medicamento que le habían recetado en la sanidad pública, por que no se lo pudo permitir por motivos económicos. Equivale a 1,9 millones de personas. No son una ni dos.
Por otra parte, la exigencia de altos precios para los nuevos medicamentos provoca problemas de acceso por retraso en la financiación pública. Así, el último informe de IQVIA para Farmaindustria, señala que entre que se aprobaba un medicamento en la UE y se decidía su financiación pública en España, había una demora de 629 días (con datos referidos a 2022).
Además, según el mismo informe, el 51% de los medicamentos aprobados tenían alguna restricción en su utilización (indicaciones, tipo de pacientes, visados de inspección, etc.).
También se detectan problemas de acceso por desabastecimiento de algunos medicamentos. Situación que, en la mayoría de los casos, está provocada por la retirada de productos a precios antiguos para sustituirlos por otros nuevos, con precios diez o cien veces más altos, cuyo valor terapéutico en muchas ocasiones es similar.
Ahora bien, si hay limitaciones de acceso en nuestro país, esas limitaciones son mucho mayores en el ámbito global, provocando gravísimos problemas de salud. Tal es así, que más de 2.000 millones de personas en el mundo, según la OMS, no pueden acceder al medicamento que necesitan. Y se estima que 10.000.000 de personas mueren cada año en el mundo cuando podían haber salvado su vida con la medicación apropiada. Por ejemplo, como denuncia Winnie Byanyima, directora de ONUSIDA, el lenacapavir para prevenir y tratar el VIH/SIDA, tiene un coste de fabricación de 44$ por tratamiento y año, y se vende a 44.000$ por tratamiento y año (15, 16). En España el precio ronda los 20.000 euros por tratamiento. Recordemos que, al mismo tiempo, mueren cada año en el mundo más de 600.000 personas por SIDA. Los monopolios en medicamentos matan. Los descubrimientos relacionados con la salud deben ponerse a disposición de todos los pacientes, sin discriminación y cuanto antes. Es una exigencia ética.
Lo vimos también en las vacunas para la COVID-19. Los países ricos, como la UE, acumulamos más de diez dosis por persona, mientras en los países del Sur Global se quedó sin vacunar más de la mitad de la población. La mayor parte de la investigación se había financiado con fondos públicos a través de subvenciones, investigación básica y compras anticipadas.
Pero los Derechos de Propiedad Intelectual se concedieron a las empresas. El coste de producción era 1 dólar por dosis. Se vendió a un promedio de 18 dólares por dosis. Las empresas, Pfizer, Moderna y otras, a través de sus asociaciones mundial y europea, se opusieron a la suspensión de patentes durante la pandemia, y los representantes de la UE y EEUU en la Organización Mundial de Comercio bloquearon el acuerdo. La falta de vacunas ocasionó varios millones de muertes evitables. A cambio, el beneficio abusivo de las empresas, por encima de los costes de fabricación, superó en dos años los 200.000 millones de dólares en venta de vacunas, medicamentos y diagnósticos para la COVID-19.
Estas enormes ganancias son la causa de que el Tratado de Pandemias, que se está discutiendo en la OMS desde 2022, no haya logrado todavía un acuerdo (la semana pasada y esta sigue discutiendo el grupo negociador internacional, y se anuncia que continuará la discusión hasta la próxima Asamblea Mundial de la Salud en mayo de 2025). Los países del Sur Global exigen que, en caso de pandemia, se suspendan los derechos de propiedad intelectual, los monopolios, y se transfiera la tecnología y el conocimiento, para poder fabricar en todo el mundo y vender a precio de coste. Los países del Norte, EEUU, la UE, Japón, Canadá, etc., presionados por la industria farmacéutica, se niegan. En cambio, estos países quieren un tratado que exija a las naciones del Sur global que compartan la información sobre patógenos y que adopten medidas de control. Puede más la codicia de unos pocos que la salud de la población mundial. Es un neo colonialismo que se está aplicando en este y en otros campos, bajo una retórica de derechos humanos y democracia.
2.3.
Un tercer problema ético que ocasiona este modelo es que, el patrocinio de la I+D por las empresas, a través de patentes y sobreprecios, provoca sesgos en la investigación (17, 18). En efecto, en la investigación patrocinada por una empresa con ánimo de lucro los sesgos de diseño, selección y análisis de los datos, y publicación de resultados, son inevitables. No olvidemos que la prioridad de la empresa es lograr un medicamento con expectativas de obtener el máximo beneficio económico posible.
Con esa finalidad, los resultados de las primeras fases de la investigación se van a publicitar, y las empresas van a conseguir que el valor de sus acciones suba, mucho antes de que esté completada la investigación y comercializado el producto, mucho antes de que sepamos si funciona o no. Las empresas patrocinadoras controlan la información a lo largo de todo el proceso. Se llama gestión de expectativas. Es decir, especulación. El profesor Joan Ramon Laporte, catedrático de farmacología de la Universidad Autónoma de Barcelona, en su libro “Crónica de una sociedad intoxicada”, explica como: “Cada nuevo fármaco es evaluado por su compañía titular, y las autoridades reguladoras dan por buenos los resultados de sus estudios, sin que exista un control efectivo sobre la veracidad de los datos recogidos. A menudo los resultados obtenidos en la investigación clínica son secretos. Si no son favorables al fármaco, no son publicados, o bien se publica una versión maquillada, o simplemente inventada. La investigación clínica publicada sobre los nuevos fármacos -concluye- es a menudo fraudulenta y casi siempre engañosa”. También, sobre este mismo tema, es recomendable la lectura del libro de Marcia Angell, “La verdad acerca de las compañías farmacéuticas. ¿Cómo nos engañan y qué podemos hacer al respecto?”.
La doctora Angell fue editora de la revista New England Journal of Medicine durante más de dos décadas. En un artículo publicado en 2009, afirmaba: “Existen conflictos de intereses y sesgos similares en prácticamente todos los campos de la medicina, particularmente en aquellos que dependen en gran medida de medicamentos o dispositivos. Simplemente ya no es posible creer gran parte de la investigación clínica que se publica”. Y en el libro citado añade: “Los ensayos clínicos puede ser amañados de muchas maneras, y esto ocurre todo el tiempo”… “Las compañías están implicadas en cada detalle de la investigación, desde el diseño del estudio, al análisis de los datos, a la decisión de publicar o no los resultados” … y concluye “Los investigadores ya no controlan los ensayos clínicos, lo hacen los patrocinadores”.
En muchos casos, la investigación realizada con financiación pública o en instituciones públicas, es aprovechada o “comprada” por las compañías farmacéuticas, en ocasiones a través de compañías start up creadas por las universidades o los centros de investigación. Una vez comprados los Derechos de Propiedad Intelectual las empresas farmacéuticas fijan precios abusivos para los medicamentos desarrollados, aunque el grueso de la investigación se hubiera realizado con dinero público directo. Un ejemplo fue el Sofosbuvir, antes citado. Esta situación se fue generalizando en EEUU con la aprobación de la Ley Bayh-Dole en 1980, y progresivamente se aplicó en Europa. Se popularizó la llamada “colaboración público privada” para lograr que la investigación “llegara” a la sociedad. El problema es que no se controló que las empresas abusaran en la fijación de precios, con lo cual, como hemos visto, muchos medicamentos no llegan realmente a quienes los necesitan.
2.4.
Un cuarto problema ético al financiar la I+D con patentes y monopolios es la fijación de prioridades de investigación. Las empresas quieren investigar en medicamentos rentables, que favorezcan sus intereses comerciales. No en necesidades de salud. Apuestan por una investigación incremental que, con pequeñas modificaciones, les permita “reverdecer” las patentes, subiendo los precios, para tratar las enfermedades crónicas prevalentes en el mundo rico (podemos citar el caso del Entresto, de Novartis, y su maraña de patentes (19). Estas grandes empresas no invierten en enfermedades de países pobres que no pueden pagar (enfermedad de Chagas, dengue, leishmaniasis, lepra, tracoma, filariasis, tuberculosis, malaria, etc.). En 2022 gastamos en I+D biomédica global, en todo el mundo, 350.000 M$ (250+100). En enfermedades
olvidadas apenas 5.000 M$, un 1,4%, cuando afectan al 20% de la población (20).
Tampoco se investiga en antibióticos, que no son rentables, aunque mueran miles de personas al año, también en Europa, por falta de antibióticos eficaces.
Como es esperable, el exceso de dinero que pagamos a la industria para I+D, tampoco se gasta en investigar en intervenciones de prevención, de promoción de la salud, de cuidados de enfermería, etc. En este sentido, la profesora Carmen Estrada, neurocientífica con más de 30 años de experiencia investigadora, afirma: “si lo que queremos es mejorar la salud de la población quizá sería más interesante plantearse como objetivo disminuir la pobreza, más que crear un nuevo medicamento”.
Sin embargo, a estas empresas no les interesa invertir en prevención de enfermedad y promoción de salud, sino hacer negocio con la cronicidad. Por eso, David Healy, profesor en Wales, en su libro “Pharmagedon” señalaba el cambio profundo en la naturaleza de los medicamentos comercializados y en la práctica de la medicina. En este escenario, dice, “las compañías farmacéuticas venden enfermedades más que curaciones”.
Además de que se fijen las prioridades de investigación por los intereses de negocio, las patentes y los secretos comerciales hacen que se dificulte la investigación cooperativa, y se retrase el acceso a resultados de investigación, obligando a repetir estudios en paralelo, desperdiciando así tiempo y recursos.
2.5.
Un quinto problema ético es que parte del exceso de beneficios destinados teóricamente a I+D, se canaliza a la formación continuada de los médicos y otros profesionales, congresos, guías clínicas, revistas, sociedades científicas, asesorías, conferencias, y un largo etc., a través de acciones de marketing. Según elEconomista, en 2021 los médicos españoles recibieron 587 M€ de la industria farmacéutica. A todos estos pagos les llaman “transferencias de valor”. Son acciones dirigidas a influir en los profesionales para inclinarles a favorecer los intereses de la empresa pagadora. Los doctores Juan Gérvas y Mercedes Pérez-Fernández, afirman que las “transferencias de valor” de las industrias farmacéuticas no crean “valor” sino que corrompen a
médicos y sociedades científicas (21).
La revista de la AAJM publicó una investigación de Ángel María Martín, en la queanalizó todos los pagos a profesionales de la salud efectuados y publicados por las 18 multinacionales farmacéuticas con mayor volumen de ventas en el año 2022 (22). El análisis muestra que 855 profesionales, líderes de opinión, habían declarado recibir más de 15.000 euros cada uno, de dichas multinacionales farmacéuticas. De ellos, 71 recibieron más de 70.000 euros anuales y nueve por encima de 100.000 euros. Esto solo de información declarada y solo de 18 empresas.
Muchos profesionales que reciben estos pagos de la industria están convencidos de que no influye en su comportamiento, ni en su investigación, ni en sus aportaciones a la guía clínica correspondiente. Pero no es así. Diversas investigaciones demuestran cómo la recepción de pagos, aunque sea tan solo la invitación a una comida, influye en las pautas de prescripción, o en la opinión expresada. El refranero, que atesora la sabiduría popular, ya lo sentencia: “es de bien nacido ser agradecido”. Otros profesionales sí son conscientes de que recibir patrocinio de la industria condiciona su investigación o su prescripción, pero preguntan: “¿Qué alternativa tengo para formarme, o para llevar adelante este proyecto? Mientras las cosas no cambien tengo que seguir dependiendo de la industria”. Precisamente por eso mismo hemos de cambiar las cosas.
Al mismo tiempo, “Las farmacéuticas destinan 110 millones de euros al año a financiar a las asociaciones europeas de pacientes”. La mayoría también aseguran que este patrocinio no condiciona sus posiciones, ni su opinión, ni socava su independencia. Sin embargo, casi todas estas asociaciones suelen apoyar los puntos de vista de la industria.
Esta influencia del patrocinio se traduce en un sobre – consumo, una “sociedad intoxicada”, como dice Laporte, presionando a prescriptores a recetar y a pacientes a consumir, y provocando efectos adversos que serían evitables: se estiman alrededor de 16.000 fallecidos anualmente en España, es decir, nueve veces los fallecidos en accidentes de tráfico cada año, y alrededor de 200.000 fallecimientos anuales en la UE, “así como decenas de miles de casos de enfermedades”, por reacciones adversas a medicamentos (23).
Para desmedicamentalizar la sociedad tenemos que empezar cada una de nosotras y de nosotros siendo cuidadosos con los fármacos que tomamos, como recuerda Peter Gotzsche, cofundador de la Cochrane Collaboration y profesor de la Universidad de Copenhague, en su libro “Medicamentos que matan y crimen organizado”: “Las compañías farmacéuticas -dice- han multiplicado sus ganancias vendiendo medicamentos a personas que no los necesitan”. Y añade: “El control de la economía de mercado en el ejercicio de la medicina no cubre demasiado bien las necesidades de los pacientes y resulta incompatible con la ética que debe regir la profesión”.
2.6.
Un sexto problema es que el exceso de pago que hacemos por I+D permite también a las empresas destinar parte de sus beneficios a lobby, para perpetuar o reforzar el sistema. Parlamentarios, gobernantes, directores generales, son tentados con las puertas giratorias (por ejemplo, en la junta directiva de Farmaindustria hay dos exdirectores generales de instituciones públicas, uno de farmacia y otro de salud pública.
En su órgano ejecutivo hay tres, de diez, que provienen de altos cargos del Ministerio de Sanidad). Las empresas también dedican parte de sus ingresos abusivos a financiar las Agencias de Evaluación, la FDA, la EMA, la Agencia Española de Medicamentos y Productos Sanitarios. No es de extrañar que la revista Prescrire encontrara que solamente 11 de los 124 medicamentos autorizados por la EMA en 2022 representaban un avance terapéutico notable para los pacientes. 23 podían ser moderadamente útiles, 76 no aportaban nada nuevo sobre los tratamientos existentes (aunque, por supuesto, se comercializarán a precios mucho más caros) y 14 eran más peligrosos que beneficiosos (24).
En la UE las compañías farmacéuticas destinaron 36 M€ para lobby en 2022, con 290 lobistas en Bruselas (25). Por su parte, en EEUU las empresas farmacéuticas destinaron 372 M$ en 2022 para lobistas en el Congreso. Un lobista por cada congresista. Así, por ejemplo, consiguieron diluir la ley que, a partir del informe Maloney antes citado, pretendía que Medicare negociase los precios de todos los medicamentos: de momento no se aplicará a todos los medicamentos, ni siquiera a 100, se aplicará solo a 10 medicamentos, y comenzando a partir de 2026. Pero, aún así, las empresas han recurrido esta decisión y tratan de anularla.
En nuestro ámbito, en la pasada legislatura del Parlamento Europeo, se ha discutido la nueva legislación farmacéutica. La propuesta inicial de la Comisión, muy moderada, planteaba por primera vez una reducción del tiempo de exclusividad, es decir, del monopolio. Se mantenía el lenguaje que lo consideraba un “incentivo”, pero se consideraba un tiempo excesivo.
También se proponía el desarrollo de una infraestructura pública europea potente para la investigación y desarrollo de medicamentos, según la iniciativa que había elaborado el STOA, el Panel para el Futuro de la Ciencia y la Tecnología del propio Parlamento. Ambas propuestas y otras más que trataban de mejorar la situación, fueron bloqueadas en el Parlamento por la mayoría conservadora. La presión de la industria es enorme.
En España también el Gobierno está revisando la legislación farmacéutica. Esperemos que sus propuestas introduzcan mejoras sustantivas de cara al desarrollo de una política farmacéutica más justa y saludable.
3.
Hasta aquí hemos analizado seis efectos adversos, no pequeños, del modelo de financiación de la I+D de nuevos medicamentos a través de monopolios, es decir, de financiación pública indirecta.
¿En qué dirección podemos avanzar?
¿Existen Alternativas para la financiación y el desarrollo de la investigación biomédica
que, a su vez, fortalezcan el SNS y beneficien al conjunto de la sociedad?
Sí que existen, aunque no es fácil desarrollarlas por la enorme resistencia al cambio.
No olvidemos que este modelo de patentes y monopolios en el medicamento es parte de un sistema económico, un nuevo capitalismo financiero global, que desde los años 80 del siglo pasado ha ido desequilibrando la distribución de los recursos en favor de las grandes corporaciones y grandes fortunas, de tal manera que hoy acumulan un inmenso poder. Según e informe de Intermon Oxfam 2024, el 1% más rico del planeta posee más riqueza que el 95% de la población mundial.
En ese contexto, como hemos visto, el modelo de monopolios en medicamentos ha degenerado exigiendo precios abusivos, y es causa de que millones de personas no tengan acceso a las vacunas y a otros medicamentos que necesitan. Pero, a su vez, produce enormes ganancias para unos pocos. Por eso es tan difícil cambiar este modelo. Pero, desde luego, es una exigencia ética, y así lo entendió el Panel de Alto Nivel sobre acceso a medicamentos convocado por la Secretaría General de Naciones Unidas en 2016, pidiendo a los gobiernos un cambio de sistema para financiar la I+D.
En palabras de Mariana Mazzucato: “Proteger la salud pública requiere un entorno para la investigación radicalmente diferente del que tenemos, con múltiples actores trabajando juntos en caminos dinámicos que compartan el conocimiento y aceleren el progreso” (26).
3.1.
En el largo plazo, por lo tanto, debemos trabajar para implantar un nuevo modelo, que beneficie sobre todo a los pacientes y al conjunto de la sociedad. Para ello se requieren dos decisiones en el ámbito global.
La primera decisión es cambiar el acuerdo ADPIC en la Organización Mundial del Comercio, para excluir a los medicamentos y productos sanitarios de la protección de las patentes y otros derechos de propiedad intelectual, como el secreto comercial. En medicamentos no debe haber ni monopolios, ni secretos.
La segunda decisión se debe tomar en la Organización Mundial de la Salud, avanzando en la propuesta que ya realizó en 2012 y quedó frenada por los países del Norte y la presión de la Big Pharma. Se trataría de aprobar un Convenio Internacional para el acceso a los medicamentos,
creando un Fondo Global para investigación y desarrollo, financiado por los países con
aportaciones proporcionales a su PIB.
-La gestión del Fondo se realizará por personas expertas designadas por los gobiernos
de todo el mundo.
-Las Prioridades de investigación se fijarán según necesidades de salud.
-La Investigación será abierta y cooperativa.
-El resultado de la investigación tendrá licencias abiertas, no exclusivas.
-Se distribuirá la información, el conocimiento y la tecnología necesaria para fabricar el
producto.
-De esta forma se podrá llevar a cabo la fabricación en todo el mundo.
-Y el precio de venta de los productos será el precio de coste.
¿Se puede financiar este modelo?
En el mundo se han gastado en medicamentos 1,6 billones de dólares en 2023. Si todos los medicamentos se hubieran vendido a precio de genérico o biosimilar el gasto hubiera sido de 400.000 millones de dólares.
A su vez, el gasto en I+D de las empresas fue de 250.000 millones de dólares.
Quiere decir que el beneficio excesivo que gastaron los países y los pacientes fue de 950.000 millones de dólares, que podrían ahorrarse y destinarse a otras finalidades. El modelo alternativo de I+D no solo se puede financiar, sino que recuperarían ingentes recursos para otros programas de salud.
El Instituto de Salud Carlos III en España, los Institutos Nacionales de Salud en EEUU, o los Programas de investigación en la Unión Europea contienen elementos de lo que podría ser un nuevo modelo de financiación y desarrollo de la I+D a nivel global. Hay que avanzar en esa dirección.
3.2.
Ahora bien, mientras tanto se pueden y deben impulsar iniciativas en el ámbito europeo y en el ámbito nacional.
3.2.1.
En el ámbito europeo.
-Limitar la duración de los monopolios al tiempo en que se recuperen los gastos de I+D
debidamente acreditados.
-Financiar la I+D realizada por empresas, con contratos de “compra anticipada” o similar, reteniendo los Derechos de Propiedad Intelectual en la titularidad pública, para así poder hacer transferencia de la tecnología y fijar precios en relación con el
coste.
-Fomentar la Investigación y el desarrollo de medicamentos con financiación pública directa con la creación de una Plataforma pública de investigación y desarrollo en la UE, como propuso STOA. Entre tanto, fortalecer la estructura de la Autoridad europea para la preparación y respuesta ante pandemias, de forma que asegure el desarrollo, producción y distribución de medicamentos, vacunas y otras contra-medidas en situaciones de emergencia.
-En todos los proyectos, becas, subvenciones, desgravaciones, etc., que tengan financiación pública de la UE fijar condicionalidades para garantizar el acceso, la asequibilidad, la investigación abierta y la transferencia de conocimiento (27).
3.2.2.
En el ámbito nacional.
Hay muchas medidas que se pueden y deben tomar a corto plazo. Señalaré solo algunas.
-Modificar la Ley Medicamento, para tratar que los precios tiendan a aproximarse a los costes de fabricación y de I+D, exigiendo toda la información sobre costes de producción y de I+D, acreditada y supervisada, y negociando precios coste-plus.
-Potenciar la acción de las Autoridades de la Competencia para vigilar el abuso de posición dominante, así como la formación de cárteles en empresas de genéricos y biosimilares. Y, en casos de importancia mayor para la salud, o de mayor impacto presupuestario, aplicar licencias obligatorias.
-Consolidar y mejorar la financiación pública directa de la investigación biomédica, a través del ISCIII y sus redes.
-Desarrollar Empresas Públicas para la fabricación de medicamentos, y potenciar las instituciones públicas para el desarrollo de vacunas y otros productos de diagnóstico y tratamiento.
-Apoyar el desarrollo de CAR-T académicas públicas, a través de la Red Española de Terapias Avanzadas, TERAV. Como señala su coordinador, José María Moraleda, se trata de acercar el tratamiento al paciente, dar un impulso científico a la sanidad pública y abaratar el precio: cuatro veces menos que el producto comercial.
-Asegurar la financiación pública de la Formación continuada de los profesionales sanitarios, de las Sociedades Científicas y de sus Publicaciones, así como de las Asociaciones de pacientes.
-Desarrollar programas potentes, sin patrocinio privado, de Evaluación de efectos adversos de los medicamentos. Y publicitar los hallazgos.
-Impulsar programas de de-prescripción. Sobre todo, en personas mayores, y sobre
todo, en residencias colectivas.
-Asegurar financiación pública para todas las guías clínicas que se utilicen por profesionales del SNS.
-Y Realizar campañas de concienciación social sobre el consumo responsable, así como sobre el abuso de las empresas farmacéuticas en los sobre precios.
Entre otras muchas
4.
Conclusión.
La financiación pública directa de la investigación biomédica es determinante y genera la mayor parte de la innovación, aunque las empresas farmacéuticas controlan la mayor parte de los resultados y la explotación de estos.
La financiación de la Investigación en medicamentos a través de monopolios perjudica al Sistema Nacional de Salud y produce una serie de efectos adversos con implicaciones éticas graves.
Los gobiernos deben adoptar medidas que garanticen y aumenten la financiación pública directa de la investigación biomédica, y deben desligar progresivamente la fijación de los precios de los medicamentos, de la financiación de la I+D.
Como recuerda la profesora Carmen Estrada, la mayoría de los investigadores han aportado sus conocimientos a lo largo de la historia sin exigir un monopolio para lograr enriquecerse. Han compartido, y comparten su conocimiento para contribuir al progreso de la humanidad.
No todas las soluciones dependen de los demás. Cada una de nosotras y de nosotros puede ser un factor de cambio. Nuestras decisiones son relevantes. Especialmente las que podéis llevar a cabo desde los Comités de Ética de la Investigación biomédica.
Por todo lo expuesto, y con la evidencia disponible, me atrevo a afirmar que es posible mantener y mejorar la investigación en nuevos medicamentos reforzando al mismo tiempo la independencia y viabilidad del Sistema Nacional de Salud.
Para finalizar, recordaré que cuando a mediados del pasado siglo, el conocido periodista de la CBS Edward Murrow le preguntó a Jonas Salk, descubridor de la vacuna contra la polio, de quién era la patente, éste se sorprendió, y tardó unos segundos en contestar. Luego dijo: “De la gente, diría yo. ¿Podrías patentar el sol?” (28, 29).
Referencias
(1). Informe El Sistema Sanitario: situación actual y perspectivas de futuro. Consejo
En 2003, dos periódicos de tirada nacional, EL PAÍS (aquí) y EXPANSIÓN (aquí y aquí) publicaban sendos artículos sobre el gasto en farmacia de receta con un título casi idéntico y en el que se tildaba a dicho gasto de «despilfarro». El de EXPANSIÓN, además, señalaba claramente un culpable: «La responsabilidad de tanto dispendio no es del consumidor, sino del médico que es el que receta«. Aunque el consumidor tampoco salía muy bien parado si nos fijamos en la viñeta acompañante del artículo y que se adjunta («De lo que no vale nada, buena sartenada«). Sin embargo, jamás en la vida he leído en ningún medio nada parecido referido al gasto público en farmacia hospitalaria. Ningún medio osa decir que el gasto en farmacia hospitalaria es un «despilfarro» ni acusa al médico hospitalario de ningún «dispendio». Veamos la evolución de ambos gastos desde 1995 hasta 2023.
Procedencia de los datos
Hasta 2014, los datos del gasto público en farmacia hospitalaria proceden de diversas fuentes utilizadas en esta entrada de 2017 (aquí) mientras que los del gasto en farmacia de receta proceden de la Estadística del Gasto Sanitario Público del Ministerio de Sanidad (aquí). A partir de 2014, tanto los datos del gasto público en farmacia hospitalaria como los del gasto en farmacia de receta proceden del Ministerio de Hacienda (aquí).
Cuantía de ambos gastos
La siguiente figura muestra la evolución de la cuantía (millones de euros) del gasto público en farmacia hospitalaria y del gasto en farmacia de receta.
La siguiente figura muestra el gasto público en farmacia hospitalaria como porcentaje del gasto en farmacia de receta.
Distribución del gasto farmacéutico público total
La siguiente figura muestra la distribución porcentual del gasto farmacéutico público total entre la farmacia hospitalaria y la farmacia de receta.
Crecimiento acumulado
La siguiente figura muestra el crecimiento acumulado del gasto público en farmacia hospitalaria y en farmacia de receta.
Comentario
Entre 1995 y 2023, el gasto público en farmacia hospitalaria se ha multiplicado por 10 mientras que el gasto público en farmacia de receta se ha triplicado. Algunas medidas tomadas durante los peores años de la crisis de 2008, (p.e., exclusión de medicamentos de la financiación pública, modificación y extensión del copago, reducción de precios y márgenes para farmacias y distribución) consiguieron la reducción del gasto público en farmacia de receta. Sin embargo. el gasto público en farmacia hospitalaria siguió creciendo incluso durante esos años de recortes. Es más, entre 1995 y 2007, ambos gastos crecieron en intensidad similar. De hecho, entre 1995 y 2007, el gasto en farmacia hospitalaria como porcentaje del gasto en farmacia de receta se mantiene estable entre el 22 y el 25%. Pero a partir de 2008, coincidiendo con el inicio de la crisis, el gasto público en farmacia hospitalaria inicia su particular escalada alcista, intensificada a partir de 2014. Así, el gasto en farmacia hospitalaria como porcentaje del gasto en farmacia de receta alcanza en 2023 el 71%. También en estos 10 últimos años, la farmacia hospitalaria ha pasado de significar el 31% del total del gasto farmacéutico público en 2013 al 41% en 2023, prácticamente ha ido sumando un punto porcentual cada año. A esta marcha, en 10-12 años podría alcanzar o superar el 50% y se equipararía o, incluso, superaría la cuantía del gasto en farmacia de receta.
Predecir el futuro es arriesgado, pero si ambos gastos mantienen su respectivo crecimiento anual promedio del periodo 2014-2023, es muy probable que en 10-12 años el gasto en farmacia hospitalaria supere la cuantía del gasto en farmacia de receta y cada uno de ellos se sitúe por encima de los 20.000 millones de euros (más de 40.000 millones la suma de los dos). Como señalaba al principio de esta entrada, parece que el gasto farmacéutico solamente es «despilfarro» si se produce en el centro de salud y no en el hospital. Cuando el gasto público en farmacia hospitalaria supere en 10-12 años la cuantía del gasto en farmacia de receta, ¿tildarán EL PAÍS y EXPANSIÓN dicho gasto de «despilfarro»?
Este texto es de un singular de interés, pues comentando el anuncio de Gilead de su licencia voluntaria para lenacapavir desmonta e ilustra muy bien el comportamiento de la industria farmacéutica, con la utilización engañosa y fraudulenta de las licencias voluntarias. Muestra como su utilización en negociaciones a puerta cerrada, selecciona países y empresas proveedores de genéricos, de acuerdo con sus intereses y estrategias comerciales debilitando seriamente la capacidad de fabricación local.
Como destaca el artículo este tiempo de estrategias de la industria farmacéutica, son en realidad una trampa que sin duda demuestra que:” No podemos confiar a las empresas farmacéuticas la responsabilidad de garantizar un acceso equitativo a los medicamentos”.
El anuncio de Gilead de su licencia voluntaria para lenacapavir, un método innovador y de prevención del VIH de acción prolongada, destaca por qué no podemos confiar en las compañías farmacéuticas para ofrecer estrategias genuinas de acceso global para los medicamentos, vacunas y diagnósticos que el mundo necesita.
Lenacapavir (LEN), una inyección dos veces al año, ha sido muy eficaz en ensayos clínicos. Podría prevenir millones de nuevas infecciones por el VIH, si fuera asequible y estuviera disponible en todas partes. Pero Gilead ha limitado los países que podrán acceder a versiones genéricas de LEN.
Inclusión y exclusión selectiva:
Si bien la licencia incluye varios países de bajos y medianos ingresos, excluye estratégicamente a muchos otros como Argelia, Argentina, Brasil, Colombia, El Salvador, Guatemala, Perú, Malasia… Solo en Brasil, 51.000 personas se infectaron recientemente con el VIH en 2023.
La licencia voluntaria (VL) de Gilead impedirá que millones de personas, especialmente poblaciones clave, accedan a LEN. Desplazará la carga de las nuevas infecciones por el VIH a los países de ingresos medios que están excluidos de la VL, que deben pagar lo que Gilead decida cobrar por el LEN. En los Estados Unidos (donde LEN está actualmente aprobado como parte del tratamiento para el VIH multirresistente), el LEN tiene un precio de 42.500 dólares estadounidenses por persona al año (PPPY). [1] Sin embargo, los expertos estiman que las versiones genéricas de LEN se pueden producir en masa de manera rentable por menos de 100 $ por persona PPPY, disminuyendo a 41 $ PPPY, una vez que el volumen alcanza los 10 millones de cursos de tratamiento por año. [2]
El acuerdo impide el suministro de genéricos a países donde no se han presentado ni concedido patentes como Argelia, Líbano, Jordania… Esto expone aún más esta licencia como una herramienta para que Gilead controle nuevos mercados e imponga su propia agenda monopolística en lugar de abordar las necesidades de salud globales.
Reforzando Las Desigualdades, No Resolviéndolas:
Esta licencia perpetúa las desigualdades al priorizar las ganancias sobre las personas. Las licencias voluntarias son controladas por la industria farmacéutica y negociadas a puerta cerrada. Las VL ofrecen acceso solo a mercados seleccionados, donde las corporaciones farmacéuticas cobran regalías y buena publicidad, mientras mantienen el control y dictan los términos de suministro en estos y en todos los demás mercados. Esta estrategia restringe el acceso y bloquea a los países en la dependencia de la supuesta buena voluntad de las empresas farmacéuticas, lo que en última instancia previene la adopción de medidas más sostenibles, como el uso de las flexibilidades de TRIPS para abordar emergencias de salud pública.
Estrategia de Pharma para socavar la soberanía de la salud:
Al vincular a los fabricantes genéricos a acuerdos de licencia que impiden el suministro a países donde las barreras de patentes se han abordado con éxito a través de licencias obligatorias u oposiciones a patentes, Gilead y otras compañías farmacéuticas socavan efectivamente el uso de las flexibilidades de TRIPS que fueron diseñadas específicamente para abordar los desafíos de salud pública. Este enfoque impide que los países aprovechen estas herramientas para producir o importar medicamentos asequibles y desarrollar sus propias capacidades de fabricación.
Bloqueo de la capacidad de producción local:
Al otorgar licencias a un grupo selecto de fabricantes, Gilead está promoviendo la dependencia continua de la producción extranjera, especialmente en las regiones de África y Europa del Este y de Asia Central. Este enfoque sofoca el crecimiento de la capacidad de fabricación local, socava los esfuerzos para lograr la autosuficiencia regional y obstaculiza la sostenibilidad a largo plazo. Estos países deben estar facultados para producir sus propios medicamentos, sin estar restringidos por acuerdos exclusivos que promuevan la dependencia de fabricantes genéricos seleccionados y limiten la capacidad de producción local.
La necesidad de soluciones de acceso global verdaderas:
Las empresas farmacéuticas, incluida Gilead, han demostrado repetidamente que no se puede confiar en ellas para liderar los esfuerzos de acceso global. La historia ha demostrado que las licencias voluntarias se utilizan como estrategia para mantener una percepción pública favorable, al tiempo que se priorizan los intereses comerciales de las corporaciones farmacéuticas. El verdadero acceso global no se puede lograr a través de esquemas de licencias fragmentados dirigidos por la empresa que continúan anteponer las ganancias a la salud de las personas.
Una llamada al cambio
Este anuncio subraya por qué necesitamos una reforma sistémica en la gobernanza mundial de la salud. Los gobiernos, la sociedad civil y los organismos y donantes internacionales deben presionar por políticas que prioricen las necesidades de salud de las personas sobre los beneficios corporativos. No podemos confiar a las empresas farmacéuticas la responsabilidad de garantizar un acceso equitativo a los medicamentos. Es hora de que los países recuperen su soberanía en materia de política de salud y utilicen todas las herramientas disponibles, incluidas las flexibilidades de los TRIPS y otras medidas, para garantizar el acceso para todos.
Este artículo de Baker, como siempre es magnífico. Estructurado desde la perspectiva en la que trabaja, que es los Estados Unidos. Aquí desarrolla su idea de financiación pública directa de la investigación, para de esta manera conseguir la reducción total de los precios abusivos generados por el sistema actual de investigación de nuevos fármacos de las farmacéuticas basado en las patentes y el monopolio.
Recomendamos su lectura completa pues sus párrafos están llenos de planteamientos y sugerencias.
La semana pasada vi a un hombre vestido de manera un tanto desaliñada en la farmacia tratando de conseguir una dosis de refuerzo contra la COVID. Le dijeron que costaría 130 dólares (era la dosis de refuerzo de Moderna). Dijo que no tenía ese dinero. El farmacéutico y un par de personas que estaban en la cola le sugirieron algunos lugares donde podría conseguirla a un coste menor, o incluso gratis. El hombre se fue y, con suerte, debió conseguir una vacuna asequible.
Recordé este incidente cuando leí un artículo del New York Times sobre los administradores de beneficios farmacéuticos (PBM, por sus siglas en inglés). La esencia del artículo es que los PBM a menudo participan en prácticas sórdidas que implican exprimir a algunas farmacias, mientras que compensan en exceso a las cadenas con las que están afiliadas. El resultado son precios más altos y un servicio de menor calidad, ya que muchas farmacias se ven obligadas a cerrar.
Hace años, Ronald Reagan decía por ahí que no necesitamos que el gobierno solucione el problema, el gobierno es el problema. En el caso del alto costo de los medicamentos, las vacunas y otros productos farmacéuticos, Reagan está en lo cierto. Los problemas que enfrentan las personas para obtener los medicamentos que necesitan a precios asequibles se deben casi en su totalidad a los monopolios de patentes concedidos por el gobierno y las protecciones relacionadas.
El punto, que sé que repito una y otra vez, es que los medicamentos son casi siempre baratos de fabricar y distribuir. En un mercado libre sería raro que un medicamento se vendiera a más de 30 dólares por receta, y a menudo por mucho menos. No tendríamos problemas para pagar nuestros medicamentos y vacunas si se vendieran a precios de mercado libre. Y no existirían los PBM en un mercado libre. ¿Tenemos “administradores de beneficios en los supermercados”?
Los monopolios de patentes otorgados por el gobierno crean este problema totalmente evitable: las personas tienen que luchar para pagar los medicamentos que necesitan para proteger su salud y posiblemente su vida. Estos pueden costar decenas de miles o incluso cientos de miles de dólares al año. Incluso si logran que una aseguradora, el gobierno o una página de GoFundMe cubran el costo, ¿por qué queremos obligar a personas que luchan con problemas de salud graves a tener además que realizar este esfuerzo?
Financiación pública: una mejor opción que los monopolios de patentes
La razón de ser de los monopolios de patentes es que son necesarios para que la industria recupere los costos de investigación involucrados en el desarrollo de nuevos medicamentos o vacunas. Si gastaran cientos de millones de dólares en desarrollar un medicamento y luego los competidores genéricos pudieran comenzar a producirlo el día que fuera aprobado por la Administración de Alimentos y Medicamentos (FDA), no tendrían la capacidad de recuperar el dinero que habían invertido. Si esta fuera la situación a la que se enfrentaban las compañías farmacéuticas, nunca invertirían mucho dinero en el desarrollo de nuevos medicamentos, ya que no sería rentable.
Este argumento es completamente cierto, pero el problema con esta lógica es que tenemos otros mecanismos para financiar la investigación necesaria para desarrollar nuevos medicamentos. Podríamos tener financiación pública. Esto no es un secreto. Actualmente gastamos más de 50 mil millones de dólares al año para financiar la investigación biomédica a través de los Institutos Nacionales de Salud (NIH) y otras agencias gubernamentales.
Si quisiéramos reemplazar la investigación respaldada por patentes que actualmente realiza la industria farmacéutica, necesitaríamos aumentar esta cantidad en alrededor de 120 mil millones de dólares al año. Eso puede parecer mucho dinero, pero probablemente ahorraríamos más de 500 mil millones de dólares al año al tener todos los medicamentos y productos farmacéuticos vendidos en un mercado libre.
Si tuviéramos que aumentar la financiación gubernamental lo suficiente para reemplazar la investigación respaldada por patentes que actualmente realiza la industria, probablemente querríamos un mecanismo diferente. Mi ruta preferida, que analizo en mi libro Rigged (es gratuito ) es un sistema en el que el gobierno firma contratos a largo plazo (por ejemplo, 10-15 años) con las compañías farmacéuticas para apoyar la investigación en un área específica.
Por ejemplo, una empresa podría contratar 40.000 millones de dólares para realizar investigaciones durante los próximos 12 años para desarrollar tratamientos y/o curas para enfermedades cardíacas. Otra empresa podría contratar para realizar investigaciones sobre cáncer de mama o cáncer de pulmón. La idea sería que el gobierno hiciera la adjudicación inicial y luego adoptara un enfoque en gran medida de no intervención, únicamente auditorías periódicas simplemente para asegurarse de que el trabajo se está haciendo realmente y que los ejecutivos de la empresa no se han ido a las Bermudas.
Puse al Departamento de Defensa como modelo para este tipo de contratación. Si bien hay muchos abusos en la contratación militar, el hecho es que al final obtenemos buenas armas.
Y tenemos una enorme ventaja con la investigación biomédica sobre la adquisición militar. Hay motivos legítimos para el secreto en la investigación militar. No queremos poner los planes para nuestros últimos sistemas de armas en la web donde ISIS pueda descargarlos. No hay base para temores similares con la investigación biomédica. De hecho, deberíamos querer que los resultados de las investigaciones se compartieran lo más ampliamente posible, para que los investigadores de todo el mundo puedan beneficiarse de los últimos avances (más sobre esto en un momento).
De hecho, podemos buscar un mejor ejemplo de contratación gubernamental exitosa que la investigación militar directa. El ex economista de la administración Biden, Ernie Tedeschi, me señaló el ejemplo de SpaceX, que ha logrado enormes avances en la mejora de la eficiencia en la puesta en órbita de objetos. A pesar del desprecio que parece tener su director ejecutivo hacia el gobierno, los logros de Musk en esta área se hicieron a expensas del gobierno. Es razonable suponer que, si una agencia gubernamental puede encontrar con éxito una empresa innovadora como SpaceX para desarrollar nuevos sistemas de cohetes, también puede encontrar empresas innovadoras para realizar una buena investigación en el desarrollo de nuevos medicamentos.
La ausencia de secreto debería ayudar en este proceso. De hecho, cuando se contratará para el desarrollo de nuevos medicamentos, una condición para obtener el dinero debería ser que cualquier contratista, así como todos los subcontratistas (la mayoría de los contratos de defensa involucran a muchos subcontratistas), publiquen todos sus hallazgos en la web lo antes posible.
Esto permitirá una rápida difusión, de modo que otros investigadores puedan aprovechar rápidamente los éxitos y aprender de los fracasos, y también limitará las oportunidades de despilfarro y fraude. Si una empresa con un contrato importante para la investigación en un área específica no tiene nada que mostrar después de seis meses o un año, será muy evidente para los expertos en ese campo compro que algo está muy mal. Si no hay una muy buena explicación de por qué la empresa no parece estar haciendo progresos, o incluso tiene fallas en la información, entonces presumiblemente perdería su contrato.
Este tipo de estrategia requeriría algún acuerdo para compartir los costos de la investigación entre países. Algunos pueden ver este tipo de acuerdo internacional como imposible, pero de hecho es exactamente lo que Estados Unidos ha estado negociando con las disposiciones del Acuerdo sobre los ADPIC de la OMC y muchas otras disposiciones sobre propiedad intelectual en los acuerdos comerciales durante las últimas cuatro décadas. Llegar a un acuerdo puede resultar polémico, pero las negociaciones sobre cuestiones de propiedad intelectual ya lo son.
Hay otra ventaja muy importante de la investigación abierta que es necesario destacar. El sistema de financiación del monopolio de las patentes ofrece a las compañías farmacéuticas un enorme incentivo para falsear la seguridad y eficacia de sus medicamentos. Los sobreprecios de los medicamentos protegidos por patentes suelen ser varios miles por ciento superiores a los costes de producción.
Esto da a las compañías farmacéuticas un enorme incentivo para promocionar sus medicamentos lo más ampliamente posible. La crisis de los opiáceos fue el ejemplo más extremo de este tipo de falsedad, en el que las compañías farmacéuticas mintieron sobre la adicción a la nueva generación de opiáceos, pero el problema surge constantemente. La FDA intenta vigilar a la industria, y unas normas más estrictas sobre la disponibilidad de datos de ensayos clínicos dificultan el engaño, pero cuando una empresa se enfrenta a ganancias tan grandes por mentir, será difícil garantizar que la evidencia sobre la seguridad y la eficacia se presente de forma completa y precisa.
En un sistema de investigación abierto, en el que los contratos se renuevan y amplían en función de una evaluación global del valor de la calidad de la investigación, habría pocos incentivos u oportunidades para mentir de una manera que pudiera tener consecuencias adversas para la salud. Las empresas intentarán presentar sus investigaciones de la mejor manera posible. Pero, en el peor de los casos, cualquier exageración conduciría a una mala asignación de los fondos de investigación, en la que una empresa menos eficaz obtendría financiación en lugar de otra que sería más eficaz. Eso es lamentable, queremos que el dinero vaya a donde sea más productivo, pero eso tiene mucha menos importancia que tratar a las personas con un medicamento que es ineficaz o incluso dañino.
Hay otro aspecto importante de este modo de financiación pública directa como alternativa a la investigación financiada por patentes. A menudo hay factores nutricionales o ambientales (por ejemplo, la exposición al plomo) que tienen efectos importantes sobre los resultados de salud. En el sistema actual, la industria farmacéutica no tiene incentivos para examinar estas posibilidades. Sólo se les recompensa por desarrollar un producto patentable.
Descubrir que el azúcar procesado puede aumentar la frecuencia de ciertos tipos de enfermedades mentales no les va a reportar dinero. Por lo tanto, no tienen incentivos para investigar este tipo de cuestiones e incluso si sus esfuerzos por desarrollar un producto patentable pudieran apuntar en esa dirección, no tendrían incentivos para compartir esa información.
Por el contrario, si se les otorgan contratos a las empresas en función de su historial de producción de investigaciones útiles, tendrían un enorme incentivo para seguir pistas que sugieran que la nutrición, el ejercicio u otros factores tienen un impacto significativo en la salud en áreas específicas. Esto podría conducir a un enfoque mucho más integrado de la salud pública.
Como llegar desde aquí hasta allá
He estado en Washington el tiempo suficiente para saber que no estamos dispuestos a reemplazar el mecanismo de financiación del desarrollo de medicamentos de una sola vez. Eso parecería enormemente arriesgado tanto desde el punto de vista económico (la industria farmacéutica es enormemente poderosa ) como desde el punto de vista sanitario. No queremos correr el riesgo de que muchos medicamentos importantes no se desarrollen porque hemos dinamitado nuestra industria.
Pero es posible imaginar un camino gradual en el que demostremos la eficacia del modelo de financiación directa en dos o tres áreas. Esto podría significar una asignación adicional de fondos al NIH con la idea de que se destinarían directamente a apoyar el desarrollo y la prueba de nuevos medicamentos, que luego estarían disponibles como genéricos desde el día en que se aprobaran.
Por ejemplo, podríamos asignar 30.000 millones de dólares durante la próxima década (3.000 millones de dólares al año) para apoyar el desarrollo de nuevos medicamentos para tratar un tipo específico de cáncer o diabetes. Esto no impediría que la industria realice investigaciones respaldadas por patentes en la misma área. Las empresas se enfrentarían al riesgo de que, si desarrollaran un nuevo medicamento, éste pudiera competir con otro que fuera igual de eficaz y se vendiera por menos de una décima parte del precio.
De hecho, ya existe una prueba de esta idea que podemos señalar. Los doctores Peter Hotez y Maria Elena Bottazzi, junto con sus colegas del Baylor College of Medicine y el Texas Children’s Hospital, desarrollaron una vacuna contra la COVID-19, Corbevax. Esta vacuna ya se ha administrado a más de 100 millones de personas en la India e Indonesia, protegiéndolas de enfermedades graves y de la muerte por COVID-19.
Corbevax se desarrolló según un modelo de código abierto. Esto significa que el proceso de producción de la vacuna, así como los datos sobre seguridad y eficacia, están completamente abiertos y disponibles para cualquiera. Eso significa que cualquier persona en el mundo con las instalaciones de fabricación necesarias puede producir la vacuna. Como resultado, la vacuna es barata: se vende a unos 2,50 dólares la dosis en la India e Indonesia.
Sería deseable que la vacuna Corbevax estuviera disponible en Estados Unidos. Aunque probablemente costaría algo más aquí, debido a los mayores costos de mano de obra y otros elementos; probablemente estemos hablando de unos 5 dólares por inyección. Eso si se compara con los 130 dólares que mi farmacia iba a cobrarle al hombre mal vestido por la dosis de refuerzo de Moderna es mínimo. (La mayoría de la gente no ve este precio, ya que las aseguradoras o el gobierno pagan gran parte o la totalidad de la factura de las dosis de refuerzo, pero en última instancia pagamos este costo de un bolsillo u otro).
Sería un gran primer paso si la FDA aprobara la distribución de Corbevax en Estados Unidos. Además de ahorrar miles de millones de dólares en el pago de las dosis de refuerzo y hacer que sean universalmente accesibles, ayudaría a dejar en claro el punto básico: los medicamentos son baratos, los monopolios de patentes otorgados por el gobierno los hacen caros. Una vez que la gente comprenda plenamente este hecho, podremos tener discusiones más inteligentes sobre el mejor mecanismo para financiar la investigación.
Uso de cookies
Este sitio web utiliza cookies para que usted tenga la mejor experiencia de usuario. Más info