Asesor sénior de Políticas en Proyectos Europeos y coordinador las relaciones con la Organización Mundial de la Salud (OMS) en el área de Acceso a Medicamentos para Health Action International (HAI).
EDITORIAL. Revista Nº 33 Octubre 2024.
¿A qué normalidad se regresa tras un episodio traumático? ¿Es acaso posible retornar a lo que antaño fue rutina y costumbre? ¿De qué manera las lecciones aprendidas o las pérdidas sufridas se incorporan a este nuevo caminar? He aquí algunas de las preguntas que podrían caracterizar las discusiones y deliberaciones que marcan la agenda de la Salud Global.
En el centro, los Estados soberanos que siguen siendo el actor principal tanto en negociaciones de presuntos nuevos tratados como en contribuciones prometidas a presupuestos y colaboraciones necesarias para el bien común. Todo ello en el marco de unas sociedades nacionales donde la desinformación constante y la manipulación rampante han convertido elementos tan inocuos (y necesarios) como vacunas, sostenibilidad y equidad en armas arrojadizas en redes y palestras.
Y ese en este contexto donde dos procesos, aparentemente inconexos, reflejan tanto las dificultades como las oportunidades de unos tiempos no por menos convulsos más favorables al acuerdo. En primer lugar, las negociaciones aun en curso (en ciernes la sesión décimo segunda) para un acuerdo o instrumento legal de carácter internacional para la prevención y respuesta a las pandemias (conocido como tratado pandémico), en segundo, las deliberaciones para la declaración política de la reunión de Alto Nivel sobre Resistencia AntImicrobial (ARM) en los márgenes de la Asamblea General de Naciones Unidas a finales de septiembre.
En ambos casos encontramos un supuesto consenso inicial alimentado en gran medida por una retórica grandilocuente sobre amenazas compartidas, a continuación, se generan unas expectativas acerca de posibles resultados en el medio y largo plazo, finalmente las discusiones se ralentizan cuando las prioridades difieren especialmente entre países ricos y empobrecidos. El actual estado de las negociaciones del acuerdo pandémico, con un creciente número de voces expresando escepticismo, y el resultado final de la declaración sobre RAM (muy lejos de los de los primeros borradores y compromisos refrendados en 2016) ejemplifican tal dinámica. La falta de transparencia en deliberaciones no contribuye a mejorar los resultados.
No existe una agenda global que vaya más allá del maximalismo de no repetición como no hay tampoco una voluntad política compartida para la colaboración y cooperación que se imponga sobre las voluntades políticas nacionales y/o regionales, alimentadas por la memoria de un acceso desigual a tecnologías sanitarias perpetrado por aquellos que hoy claman por el acceso irrestricto a patógenos. De igual manera, la llamada epidemia silenciosa de RAM afecta es cierto a Norte y Sur pero son los países en desarrollo quienes deben cargar con las tareas de análisis y prevención sin que los recursos para ello hayan sido localizados o prometidos. Aun así, hay razones para el optimismo. Si bien la actual arquitectura institucional no puede responder a los desafíos que para la Salud Global plantea un planeta profundamente desigual, no es menos cierto que lentamente se van consolidando nuevas formas de entender la movilización de recursos y la identificación de intereses comunes y compartidos; a escala local, regional y global aquellas voces que pretenden poner la salud de las multitudes por delante del beneficio de las minorías van ganando peso. En un futuro no muy lejano sus propuestas llegaran a los órganos de deliberación y decisión.
Especialista en Farmacia Hospitalaria. Servicio de Farmacia del Hospital Universitario Puerto Real.
EDITORIAL. Revista Nº 33 Octubre 2024.
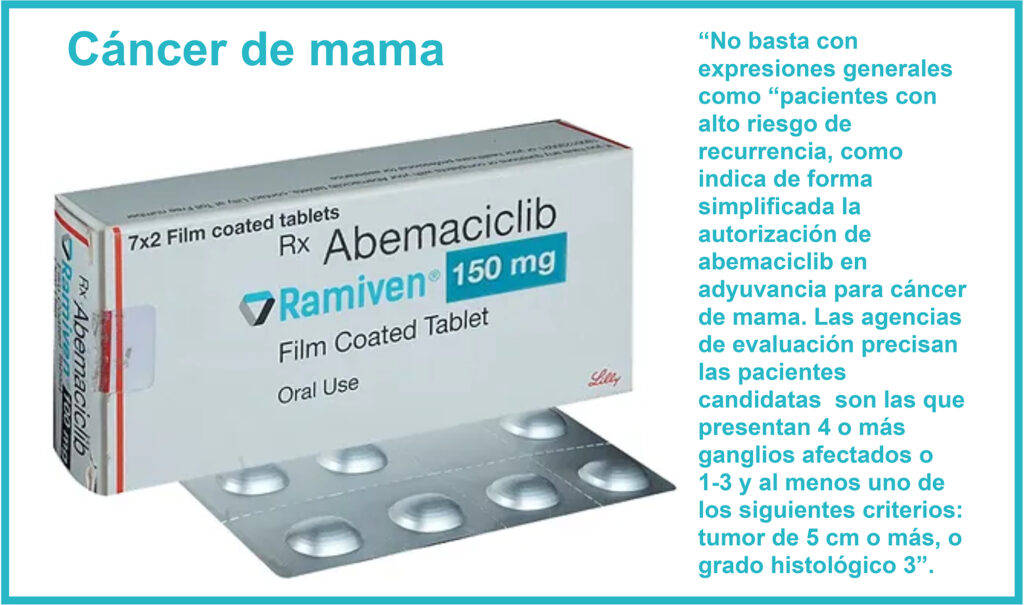
Al evaluar un nuevo medicamento para su uso clínico, es crucial identificar a los pacientes que realmente se beneficiarán de su empleo (1). Para ello, no basta con expresiones generales como “pacientes con alto riesgo de recurrencia”, como indica de forma simplificada la autorización de abemaciclib en adyuvancia para cáncer de mama (2). ¿Qué criterio debe seguirse para valorar dicho riesgo?, y más aún, ¿qué nivel de riesgo se considera “alto”? La Haute Autorité de Santé (HAS) francesa (3), el National Institute for Health and Clinical Excellence (NICE) británico, con su Patient Access Scheme (Plan de Acceso de Pacientes) (4) y los Informes de Posicionamiento Terapéutico españoles (5), dieron la misma respuesta el año pasado: las pacientes candidatas son las que presentan “4 o más ganglios afectados o 1-3 y al menos uno de los siguientes criterios: tumor de 5 cm o más, o grado histológico 3”.
Este es precisamente el valor de la evaluación post-regulatoria: no basta con señalar que abemaciclib ofrece un beneficio importante frente al tratamiento estándar. Es imprescindible especificar claramente las pacientes que pueden obtener ese beneficio para orientar la prescripción. Además, este enfoque también es clave para un correcto estudio económico (6), permitiendo estimar un impacto presupuestario realista. Si no delimitamos con precisión a quién queremos tratar, ¿cómo podríamos calcular el coste de su incorporación? Y ¿cómo tomar buenas decisiones de precio y financiación selectiva?
Preocupación ante el nuevo RD ETS
Resulta alarmante que España, siendo una de las mayores potencias científicas en evaluación de medicamentos, esté a punto de dar un paso involucionista con el nuevo el nuevo Real Decreto de Evaluación de Tecnologías Sanitarias (RD ETS) (7). Este proyecto, tal como está redactado, evita una recomendación pública y oficial sobre el escenario de utilidad del nuevo medicamento, impidiendo que el informe clínico especifique con precisión los pacientes que podrían beneficiarse. Esto se refleja tanto en los métodos como en el procedimiento de evaluación.
En lo que respecta a los métodos, el proyecto restringe drásticamente el enfoque al depender absolutamente (art. 10 L) y sin necesidad -porque no es vinculante (8)-, de la futura evaluación europea, que no tiene como objetivo especificar el escenario de utilidad. Por otra parte, el RD excluye expresamente que el informe clínico contenga “juicios sobre el posicionamiento” (art. 8.5) y ni menciona la valoración del “beneficio clínico adicional relevante” -criterio pivotal recomendado por el CAPF (9)-. Sin una clara evaluación clínica que valore la utilidad del fármaco, precisando en qué pacientes se consigue beneficio y describiendo las situaciones de incertidumbre, no habrá forma de contrarrestar la influencia de la promoción comercial, que podrá ejercer presión sobre la prescripción sin ningún contrapeso. Todo esto se hacía ya en los antiguos IPT y se recoge en el modelo de evaluación del grupo GÉNESIS (SEFH) (10), de forma que estamos ante un retroceso injustificado.
La participación de la AEMPS no es suficiente
El procedimiento consolida la carencia al especificar los pacientes candidatos, ya que el proyecto de RD delega exclusivamente a la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) la elaboración de los informes de evaluación (art. 4.3). Aunque es muy positivo contar con el rigor científico de los técnicos de la AEMPS, no es suficiente. Tampoco basta que la AEMPS contrate a técnicos externos seleccionados y dirigidos desde la propia Agencia. La evaluación clínica post-regulatoria requiere un enfoque multidisciplinar que vaya más allá de la perspectiva regulatoria.
Es importante tener en cuenta que la AEMPS, al haber participado en la elaboración de la ficha técnica del medicamento, puede enfrentarse a un conflicto interno o percibido de cara al exterior. Aunque no sea un conflicto real, la Agencia podría sentirse incómoda al detallar un escenario de utilidad más específico que el texto aprobado en la indicación. Por eso, para una auténtica evaluación post-regulatoria, es importante la participación de profesionales seleccionados por su experiencia evaluadora y clínica en los propios servicios sanitarios. Es más, esto enriquece el informe puede aportar mayor aceptabilidad de las decisiones en los propios servicios sanitarios.
En varios foros sanitarios se ha señalado que la revisión multidisciplinar de REvalMed fue un proceso lento. Aunque este modelo multidisciplinar habría necesitado ajustes y consolidación, basta revisar los Informes de Posicionamiento Terapéutico (IPT) publicados en esa época (2021-2023) para ver que muchos corresponden a fármacos con opiniones positivas del CHMP (EMA) de periodos anteriores. Con REvalMed se aceleró significativamente la producción de IPT: los nodos se activaron no solo para responder a los nuevos informes, sino también para sacar del atasco expedientes acumulados del año anterior. La publicación masiva de IPT en septiembre de 2023, atribuida erróneamente por algunos al desmantelamiento del modelo en julio (11), parece difícil de explicar como una respuesta exprés de la AEMPS en pleno agosto.
Evaluación clínica independiente, pero con especificaciones
El equipo que realiza la evaluación clínica ha de ser independiente de quienes toman decisiones sobre precio y financiación (9), evitando así que se vea condicionado por criterios económicos que puedan restringir sus recomendaciones. Sin embargo, esta independencia no debe impedir que emita una especificación clara sobre el escenario de utilidad del medicamento, sus incertidumbres y limitaciones.
¿Quién puede hacer esto mejor que un comité de profesionales capaces de valorar la evidencia científica, entenderla a la luz de su experiencia clínica y expresarla en una recomendación? Más adelante, la Comisión Interministerial de Precios se encargará de la negociación y tendrá que decidir si la propuesta clínica es financiable y si el fármaco se puede usar en todo el escenario de utilidad enunciado, o es preciso hacer alguna restricción adicional, por causa económica (1). Pero ningún responsable político se arriesgaría a tomar una decisión que podría ser percibida como restrictiva, especialmente si puede ser utilizada mediáticamente por sus opositores, sin contar con un informe científico-técnico independiente que justifique dicha decisión (12).
La importancia de definir el lugar en terapéutica: defensa de la sanidad pública
La evaluación es inútil si no hay un posicionamiento claro. Una evaluación que no valora es como los primeros y los últimos informes de posicionamiento terapéutico que no posicionan. Dejaríamos el campo totalmente libre a la influencia de la promoción comercial para ampliar su nicho de mercado, tal y como ocurría antes de que evaluación post-regulatoria se desarrollarse en las comisiones de farmacia locales. Sin un posicionamiento centralizado y riguroso, se pensaría de nuevo en implementar evaluaciones locales y autonómicas, pero estas se encontrarían deslegitimadas, al existir ya una evaluación oficial para todos, y no se podrían aplicar sus conclusiones.
Por eso, como hemos visto con el ejemplo de abemaciclib, los países con una sanidad pública avanzada han dedicado esfuerzos a establecer sistemas en los que expertos evaluadores y clínicos, con una perspectiva independiente, pueden interpretar la evidencia disponible y aportar recomendaciones claras, aterrizadas en la clínica (1). Estos sistemas son esenciales para garantizar la sostenibilidad de la sanidad pública, que debe financiar los avances terapéuticos reales sin incurrir en gastos innecesarios. Es poco probable que podamos limitar significativamente el precio que pagamos por los nuevos medicamentos a corto plazo, pero lo que sí está en nuestras manos es incorporarlos de manera razonable, destinándolos a los pacientes que realmente los necesitan.
España ha logrado construir una sanidad pública que es un referente mundial, pero sufre un deterioro evidente por diversas causas, y la escalada de precios es una de las amenazas más importantes. No podemos permitirnos dar pasos atrás. Debemos avanzar hacia una evaluación rigurosa, independiente y multidisciplinar, que se convierta en uno de los pilares para proteger nuestra sanidad pública frente a intereses comerciales y espurios, facilitando su sostenibilidad económica. Esto requiere un liderazgo político firme, tanto a nivel estatal como en las comunidades autónomas, alejándose de rivalidades políticas y considerando el enorme impacto que este nuevo RD puede tener en el futuro de nuestro sistema sanitario.
Referencias
(1). Alegre-del Rey EJ, Fénix-Caballero S, Fraga Fuentes MD, et al. La importancia del posicionamiento terapéutico en la evaluación posautorización de nuevos medicamentos. Farm Hosp. 2024 Jul 19:S1130-6343(24)00097-7. Disponible: https://www.revistafarmaciahospitalaria.es/es-pdf-S1130634324000977 [acceso 25/10/2024].
(4). National Institute for Health Care and Excelence. Abemaciclib with endocrine therapy for adjuvant treatment of hormone receptor-positive, HER2-negative, node-positive early breast cancer at high risk of recurrence. Technology appraisal guidance [TA810]. NICE; 2022. Disponible: https://www.nice.org.uk/guidance/ta810/chapter/1-Recommendations [acceso 25/10/2024].
(5). REvalMed SNS. Informe de Posicionamiento Terapéutico de abemaciclib (Verzenios®) en combinación con hormonoterapia para el tratamiento adyuvante del cáncer de mama en estadios iniciales RH positivo y HER2 negativo, con afectación ganglionar o elevado riesgo de recidiva. Ministerio de Sanidad; 2023 [Consultado 10 Oct 2023]. Disponible: https://www.aemps.gob. es/medicamentosUsoHumano/informesPublicos/docs/2023/IPT-144-Verzeniosabemaciclib.pdf [acceso 25/10/2024].
(8). “[…] el contenido del informe de evaluación clínica conjunta es de carácter científico y no debe ser vinculante […]”. Reglamento (UE) 2021/2282 del Parlamento Europeo y del Consejo de 15 de diciembre de 2021 sobre evaluación de las tecnologías sanitarias y por el que se modifica la Directiva 2011/24/UE. Diario Oficial de la Unión Europea L 458/5, 22/12/2021. Disponible: https://eur-lex.europa.eu/legal-content/ES/TXT/PDF/?uri=OJ:L:2021:458:FULL [acceso 25/10/2024].
Especialista en Medicina Interna, jubilada, Equipo Cesca, mpf1945@gmail.com
Revista Nº 33 Octubre 2024.
Los gérmenes en el ciclo de la vida
Los gérmenes son parte de la vida en la Tierra y, a su modo, conviven con sus “víctimas” en una forma que puede llegar a ser brutal, hasta el punto del exterminio de estas.
Pero, en general, los demás seres vivos llegan a un “entendimiento” con los gérmenes. A veces puede ser una especie de “intimidad obscena”; por ejemplo, los virus y fragmentos de virus que se insertan en el material genético humano. También los gérmenes intestinales que aseguran el suministro de la esencial vitamina K.
No todos los gérmenes causan infecciones, pero también los gérmenes infecciosos forman parte de la vida en la Tierra y las enfermedades infecciosas deberían ser vistas como un componente más del ciclo existencial de los seres vivos.
Las enfermedades infecciosas y la especie humana
En lo que respecta a la especie humana, las enfermedades infecciosas han acompañado y modelado su evolución. Buena prueba es nuestra condición de simbiontes, es decir, de seres en los que se combinan distintas especies en beneficio mutuo. Así, las células con genes humanos son una escasísima minoría en nuestro cuerpo, donde habitan miles de millones de bacterias, virus, hongos, levaduras y otros “bichos”.
Estos gérmenes son esenciales para mantener un ecosistema sano, con capacidad de supervivencia y de adaptación al entorno. Son más abundantes en lugares como el tubo digestivo (de la boca al ano), la vagina, la piel, las uñas, el ombligo, los conductos auditivos, el surco balanoprepucial y el cuero cabelludo.
En muchos casos desconocemos su función, si alguna. Por ejemplo, la mayoría de los virus del papiloma humano en la piel y la vagina no causan daño alguno, pero en ocasiones pueden dar lugar a las verrugas cutáneas (más en la mano, la axila, el cuello y en la planta del pie) y genitales, y al cáncer de cuello del útero.
Otros gérmenes tienen una acción muy beneficiosa, como las ya citadas bacterias del intestino que sintetizan vitamina K y ácido fólico. En el útero el feto vive en un medio estéril, pero afortunadamente se contagia por gérmenes en el momento de nacer, en el paso por el «canal del parto» (básicamente la vagina) e inmediatamente después al succionar los pezones para mamar.
Existe una dinámica fluctuante entre infecciones y seres humanos que se modifica por cambios en la biología de los gérmenes y en la conducta humana.
Así, en las infecciones fue determinante el desarrollo urbano, que desde hace unos 10.000 años ha ido agrupando humanos. También fue clave la domesticación de los animales y la estrecha convivencia con ellos (y sus propias y/o compartidas infecciones).
La situación se modificó por el desarrollo hace muchos siglos de la variolización y hace más de dos siglos de las vacunas, y recientemente de los antimicrobianos (antibióticos, antivirales y otros). Es también relativamente reciente (y no llega a todos los humanos) el suministro de agua potable, la depuración de las aguas residuales, la reglamentación de la higiene alimentaria, la redistribución de la riqueza, la disminución del tamaño de las familias, las mejoras de las viviendas y otras medidas económicas y sociales que en conjunto han mejorado la respuesta a las enfermedades infecciosas.
Atención clínica y salud pública
El sistema sanitario ofrece servicios personales y servicios de salud pública.
Los servicios clínicos personales se ofrecen en atención primaria (el médico de cabecera y otros profesionales que trabajan en el entorno y el ambiente de la comunidad), en atención ambulatoria de especialidades focales, en atención hospitalaria (centros en los que se puede alojar al enfermo mientras se le diagnostica y/o trata) y hay también servicios de urgencia que pueden desplazarse allá donde sea necesario.
El sistema sanitario ofrece también servicios de salud pública, para las poblaciones. Por ejemplo, lo que aseguran la higiene alimentaria, o el suministro y depuración de aguas, la habitabilidad de las viviendas, o las normas de manejo de productos peligrosos como material radioactivo, etc. Se incluyen las medidas específicas para evitar y paliar las enfermedades infecciosas, en que es clave la cooperación de los servicios de salud pública con los de atención clínica. Por ejemplo, en el estudio de brotes de gastroenteritis, o en la vacunación contra el tétanos (y otras enfermedades “vacunables”).
Todos los servicios sanitarios se coordinan para optimizar la respuesta a las necesidades individuales y generales en busca del mejor rendimiento posible con los recursos que la sociedad destina al sistema sanitario.
Casos y brotes de enfermedad infecciosa “vacunable”
En las últimas décadas, como resultado del desarrollo socioeconómico y de las campañas de vacunación infantil iniciadas en la década de los sesenta del siglo XX, ha disminuido la incidencia de las conocidas como enfermedades “vacunables”, enfermedades infecciosas de presentación habitual en la infancia como sarampión, varicela, difteria o tosferina entre otras.
De manera ocasional se han presentado brotes de estas enfermedades entre población no vacunada, por motivos sociales de inequidad en el acceso a los recursos sanitarios o por rechazo voluntario a la vacunación sistemática de la población infantil. Pero hay, también, casos y brotes en población bien vacunada.
Las vacunas poco eficientes, como la de la tosferina, parotiditis y difteria, y la menor eficacia en la inmunidad adquirida mediante la vacunación respecto a la inmunidad natural que se adquiere tras el proceso infeccioso, por ejemplo, en el sarampión, hacen que exista una bolsa de población correctamente vacunada pero susceptible de padecer la enfermedad y por ello se está registrando un aumento en la incidencia de algunas de estas enfermedades como parotiditis, tosferina, sarampión y otras.
Tal cuestión atañe al tiempo a los servicios de atención personal (para la vacunación en sí y para el diagnóstico y tratamiento temprano de los casos) y a los de salud pública (para la mejora de las vacunas, de su calendario y del acceso a las mismas).
Los firmantes hemos analizado, por ejemplo, el caso de la tosferina que resumimos en “La vacuna de la tosferina es una vacuna necesaria, pero que precisa mejora profunda y urgente. Ante los brotes de tosferina, y las muertes por tosferina, es importante tener en cuenta que el problema no son los antivacunas sino la vacuna. La vacuna de la tosferina es una vacuna ineficiente. Precisamos una mejor vacuna (que genere inmunidad perdurable y que evite el contagio y la transmisión) (1)”.
Respecto al sarampión, por ejemplo, en España el mayor brote que ha habido se dio en Sevilla, en 2011, entre población marginada, con 1.759 casos (uno mortal). Este caso sevillano es un buen ejemplo del origen de los brotes españoles, asociados más a la exclusión social que a la duda vacunal (2).
Por todo ello, para disminuir o evitar casos y brotes de enfermedades infecciosas “vacunables” se precisa de la cooperación de los servicios clínicos y los de salud pública.
Grupo de Vacunas de la Sociedad Española de Salud Pública y Administración Sanitaria (SESPAS) versus Comité de Vacunas e Inmunicaciones de la Asociación Española de Pediatría (CAV-AES)
En la aceptación de las vacunas son clave el pediatra y el médico general/de familia. La enfermera es esencial en el acto de la vacunación.
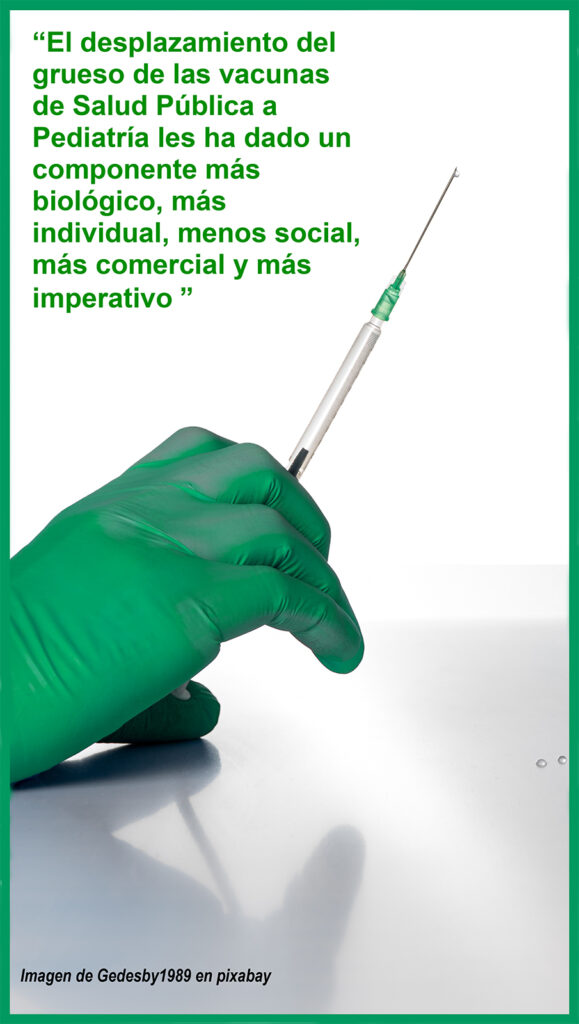
El desplazamiento del grueso de las vacunas de Salud Pública a Pediatría les ha dado un componente más biológico, más individual, menos social, más comercial y más imperativo.
Es importante recordar que la vacunación es un ejemplo primigenio de biopolítica, en el que lo deseable (conservación de la salud) se convierte en mandato (obligación moral y/o legal de ser vacunado, en este ejemplo).
Las «autoridades» (políticos, técnicos, expertos y demás) establecen un discurso y unas normas que generalmente se imponen a la sociedad, sin mucho diálogo.
Es escaso el desarrollo de una «vacunología social» en una sociedad como la española en la que se multiplican sin cesar los “expertos” en vacunas y sus grupos, se mantiene la verticalidad de las decisiones vacunales, se explota el miedo a la vulnerabilidad y se demuestra escaso o nulo interés por las valoraciones críticas de vacunados (y de sus responsables legales) y profesionales (3).
La Sociedad Española de Salud Pública y Administración Sanitaria, SESPAS, tuvo un Grupo de Vacunas dignamente coordinado por Luis Palomo (quien mantuvo y mantiene una línea de ciencia y ética acorde con los mejores intereses de la población (4)). Con el paso del tiempo, SESPAS fue perdiendo interés por las vacunas, su Grupo de Vacunas pasó a ser coordinado por José Tuells, director de la Cátedra de Vacunología Balmis Universidad de Alicante de la Fundación ASISA, y finalmente tal Grupo desapareció. En la actualidad SESPAS ignora por completo el campo de las vacunas y carece de grupo de trabajo al respecto.
Por el contrario, las industrias financiaron y financian generosamente las actividades y/o miembros de sociedades clínicas “científicas” relacionadas con las vacunas como la Asociación Española de Pediatría, AEP, y su Comité de Vacunas e Inmunizaciones (CAV-AEP), la Asociación Española de Vacunología, la Asociación Nacional de Enfermeras y Vacunas, etc.
Tenemos idea de lo que reciben algunos líderes de vacunas y las cantidades resultan obscenas, tanto del propio CAV-AEP y otros próceres (5), como de alguno en particular (Francisco Martinón, líder de opinión “diamante” que recibió 283.573 euros, declarados por las industrias en dos años, 2021 y 2022 (6)).
Conclusión
Las vacunas son medicamentos de salud pública, parte de la mejor respuesta frente a las enfermedades infecciosas.
Ante las infecciones e infestaciones conviene el uso racional de los recursos (la gestión integrada) y por ello habría que evitar el excesivo protagonismo de las asociaciones profesionales clínicas “científicas”. Tales asociaciones convierten las vacunas en respuestas individuales, no poblacionales, que sirven a los intereses de la codicia (“afán de lucro”) de los accionistas de las industrias farmacéuticas. Es decir, anteponen el lucro a la salud (“la bolsa por encima de la vida”).
La inexistencia de un Grupo de Vacunas en SESPAS es expresión simbólica del triunfo de la visión neoliberal de las vacunas como simple cuestión comercial, como negocio.
Farmacéutico, coautor junto a su hermano Raúl del ensayo “De venta de farmacias. Una denuncia del negocio de la salud desde dentro”.
Revista Nº 33 Octubre 2024.
En la justificación de motivos de la página web de la Asociación Acceso Justo al Medicamento podemos leer que, según la OMS, “el acceso equitativo a unos medicamentos seguros y asequibles es de una importancia vital para que la población goce del grado máximo de salud que se pueda lograr”.
Sin duda, es una afirmación con la que estoy totalmente de acuerdo. Sin embargo, este acceso equitativo y seguro es aplicable a todos los estratos de la asistencia sanitaria. Desde las injustas patentes de las nuevas moléculas que aparecen en el mercado, cuyos precios desorbitados privan de medicación a las personas más desfavorecidas, hasta la prescripción y dispensación de los fármacos más comunes a la población general, donde las condiciones de seguridad y equidad quedan a veces en entredicho.
Es en este último paso, la dispensación llevada a cabo en las oficinas de farmacia, donde como farmacéutico voy a hacer más hincapié. Tanto en medicamentos como en la venta (y esto es también “farmaindustria”) de tantos y tantos productos de dudosa o nula eficacia que, incluso en ocasiones, ayudan a patologizar procesos tan naturales como la menopausia o la alopecia androgénica.
Para entender a donde quiero llegar hay que saber que según la Ley 16/1997, de 25 de abril, de Regulación de Servicios de las Oficinas de Farmacia, “las oficinas de farmacia son establecimientos sanitarios privados de interés público” y añado que, como es lógico en cualquier empresa privada, su razón de ser es maximizar beneficios.
Esta definición que recoge la citada ley parece que tampoco dice mucho, pero lo quiere decir todo. Y es que, en la viabilidad de una PYME, como es la oficina de farmacia, el salario de los empleados y el beneficio empresarial dependen de la caja que entre al final del día. Esto abre la puerta a que se cometan todo tipo de tropelías que van tanto contra la equidad como contra la seguridad en el uso de los medicamentos por parte de la población.
No se me entienda mal, no digo que todos los farmacéuticos sean unos piratas ni nada por el estilo, pero sí que este modelo,) donde desde las farmacias hay dispensación de medicamentos, productos sanitarios y también complementos alimenticios, parafarmacia, etc., invita a ello.
No hay que tener mucha imaginación para saber que de las farmacias se han podido retirar medicamentos que requieren prescripción médica sin la correspondiente receta médica. Antiinflamatorios, corticoides, antidepresivos, antibióticos, diuréticos o medicamentos, que requieren una atención especial y que llevan un registro aparte como las benzodiacepinas, han sido vendidos en oficinas de farmacias con total impunidad y sin que medie control alguno por parte de la administración.
Este hecho atenta contra la equidad en el acceso al medicamento ya que, como se dice en Valencia, “amb diners, torrons” (traducido como “con dineros, turrones”), y es que muchas veces si pagas puedes obtener ese antibiótico que crees que te hace falta para el catarro leve y autolimitado que tienes, pero una persona que viva bajo el umbral de la pobreza no tiene este acceso al antibiótico sin la prescripción médica. O casos más recientes como el del principio activo liraglutida, un medicamento análogo del GLP-1 utilizado principalmente como antidiabéitco. Este es un medicamento cuyo prospecto si compramos la marca Victoza® 6mg/ml solución inyectable dice que “Ayuda a su cuerpo a reducir su nivel de azúcar en sangre únicamente cuando este nivel de azúcar está demasiado elevado”, pero que si lo compramos de la marca Saxenda® 6mg/ml solución inyectable (pongo la dosis y la forma farmacéutica para hacer ver que es exactamente igual) leeremos en el prospecto que “Saxenda® es un medicamento para perder peso que contiene el principio activo liraglutida”.
Misma dosis, misma forma farmacéutica, mismo (ojo con esto) titular de comercialización, pero diferente uso clínico. Sin embargo, nuestro sistema nacional de sanidad (SNS) no financiaba la presentación para la pérdida de peso y sí (con visado de inspección) para el tratamiento de la diabetes. Esta nueva indicación hizo que la liraglutida se vendiera como reductor de peso en el libre mercado y fuera del SNS, lo que provocó desabastecimiento del fármaco y que muchos pacientes diabéticos se quedaran sin su medicación. La venta ilícita en farmacias de este medicamento sin la correspondiente receta médica o la venta legal a cualquiera que pueda pagar a un médico privado para que le haga la receta provocaron la alta demanda de la liraglutida hasta el punto de que la cotización bursátil de Novo Nordisk subiera en los últimos 5 años un 360%. Fue tal la situación de desabastecimiento que la propia Agencia Española del Medicamento y Productos Sanitarios (AEMPS) emitió un comunicado el 8 de septiembre de 2023 con una serie de recomendaciones para paliar los problemas de suministro de los análogos GLP-1, como por ejemplo, priorizar el suministro a los pacientes diabéticos.
El caso de la liraglutida es un claro ejemplo de inequidad en el acceso al medicamento pero ya sabéis, amb diners, torrons.
Hablaba al principio de la definición del acceso justo al medicamento y no solo mencionaba el acceso equitativo sino al acceso seguro. El acceso seguro no es únicamente que el medicamento presente los menores efectos secundarios o reacciones adversas (RAM) posibles. Es además, que sea prescrito y dispensado para la patología adecuada, en la dosis mínima eficaz y durante el menor tiempo posible.
La dispensación ilícita o fuera de indicación de medicamentos sujetos a prescripción médica son ejemplos de un acceso inseguro al medicamento, pero muchas veces se hace porque “¡cómo no le vas a vender el antibiótico para su catarro! ¡Vamos a perder un cliente y encima la farmacia de al lado se lo va a vender!”. Y sí, es cierto. La farmacia puede perder un cliente (fijaos que digo cliente y no paciente), el empleado puede perder el puesto de trabajo (a un servidor le despidieron de una farmacia el primer día de trabajo por negarse a vender antibióticos sin receta) y seguramente la farmacia que está a 250 metros se lo acabe vendiendo. Y pasa, pasa mucho. Desde antibióticos a corticoides, pasando por antidepresivos, o levotiroxina (un medicamento para el hipotiroidismo que requiere una dosis muy ajustada y que puede provocar una “tormenta tiroidea”, un cuadro potencialmente fatal por exceso de hormona tiroidea). La levotiroxina se compra ilícitamente para perder peso. Al igual que los medicamentos conocidos como “diuréticos”, que engloban a multitud de moléculas con características farmacológicas diferentes y cuyo medicamento más famoso es la furosemida, conocido por su marca comercial como Seguril®.
Hubo un caso en la provincia de Valencia de muerte de un hombre a manos de su pareja por envenenamiento con laxantes y furosemida. Bien es verdad que los laxantes se pueden comprar sin receta médica y basta con recorrerse muchas farmacias comprando un par de cajas en cada una para no levantar ninguna sospecha. El caso de la furosemida es diferente, ya que es un medicamento que no puede venderse sin receta médica. Pues la ya sentenciada asesina se hizo en 3 meses con más de 2.000 pastillas de Seguril®. Cada caja tiene entre 10 (el envase pequeño) y 20 (el envase grande). Esto hace como mínimo que la mujer comprara 200 cajas de Seguril® en 3 meses. Un medicamento, recordemos, que se dispensa únicamente bajo prescripción facultativa. Ni siquiera una farmacia que venda absolutamente de todo sin receta médica, dispensaría tanto diurético a la misma clienta sin sospechar lo más mínimo y “cerrar el grifo”, lo cual nos lleva a la conclusión de que muchas farmacias de forma individualizada dispensaron la medicación de forma ilícita.
Como vemos, la seguridad en el acceso al medicamento está sujeta, en multitud de ocasiones, a la caja de una empresa privada. Como bonus track antes de dejar la dispensación de medicamentos diré que, en otra farmacia en la que trabajé, me llevé una dura reprimenda por parte de la dueña por negarme a vender a una amiga suya tres cajas de alprazolam, más conocido como Trankimazin®. Decir que, al tiempo, acabé despedido de forma improcedente porque no “me veían con futuro en esa farmacia” tras decir que me estaba preparando para hacer el FIR.
Dejemos atrás los medicamentos sujetos a prescripción médica para hablar de otro de los productos expuestos y vendidos en la oficina de farmacia. Los complementos alimenticios.
Y es que no solo de la venta de fármacos vive la oficina de farmacia. Los complementos alimenticios suponen un bocado importante en la facturación de muchas farmacias y, como todo en esta vida, no es oro todo lo que reluce y el único beneficio que aportan es a la caja de la empresa.
Complementos o suplementos alimenticios como el colágeno para las articulaciones, vitaminas para la caída del pelo, productos para fortalecer las defensas, vitamina C o jalea real para los resfriados, pastillas a con fósforo para mejorar la memoria… no solo no han sido demostrados en ensayos clínicos medianamente serios o no han pasado la evaluación de una revisión sistemática o un metaanálisis.
La venta de estos productos está asociada a las “declaraciones de propiedades saludables” o health claims por su nombre en inglés. Esto, explicado de forma rápida y sencilla, es que si un alimento o producto alimenticio presenta un nutriente o molécula que intervenga en un proceso fisiológico determinado, podemos etiquetar el producto con una declaración de propiedad saludable sobre ese proceso fisiológico en cuestión.
Dicho de otra manera y con un ejemplo: la vitamina C interviene en una reacción química que se da en nuestro organismo durante la síntesis del colágeno, promoviendo la hidroxilación de los aminoácidos lisina y prolina para fabricar hidroxilisina e hidroxiprolina, respectivamente. Estos aminoácidos seguirán su curso, junto con otros aminoácidos más, más reacciones y más moléculas que al final del todo, darán como resultado la molécula de colágeno.
Pues bien, según esta health claim podemos vender cualquier cosa que tenga vitamina C, ya sea un complemento alimenticio o un kilo de naranjas y ponerle la etiqueta “contribuye al normal funcionamiento de las articulaciones”. Pasa también con el fósforo, que está presente en la molécula fosfatidilcolina, que a su vez forma parte de las membranas celulares, entre ellas de las neuronas. Pues a raíz de esto podemos añadir una coletilla a los productos o complementos a base de pastillas o brebajes que contengan fósforo que diga “contribuye a mantener la memoria”. A pesar de que una sardina tenga más fósforo que toda una caja de pastillas.
Pues todos estos productos están al alcance de cualquiera en la oficina de farmacia y vendidos por la seriedad que da una bata y una plaquita que ponga «farmacéutico». Y, desde mi punto de vista, no es tan grave que no hagan nada de lo que prometen (o igual que si nos comemos una naranja, una sardina o unas manitas de cerdo con todo su colágeno), sino que contribuyen a la medicalización de la sociedad para síntomas banales o procesos totalmente fisiológicos y que no representan ninguna patología. Procesos fisiológicos como la menopausia o a alopecia androgénica en el varón no son enfermedades ni se van a curar con suplementos ni complementos alimenticios. Igual que no vamos a curar antes un resfriado con jalea real ni vamos a mejorar la artrosis tomando colágeno. Pero sí, nos acostumbramos a que, por cada problema, sea real, figurado o directamente inventado por (sí, esto también es industria farmacéutica a su modo) tenemos una solución a base de comprimido, ampolla, o spray que poder comprar por un módico precio en nuestra oficina de farmacia.
Así que, para concluir, no solo necesitamos una industria farmacéutica pública que investigue, desarrolle y ponga novedades farmacéuticas (útiles y que supongan un adelanto terapéutico) en circulación sin el abusivo escándalo de las patentes, sino que además necesitamos una distribución y dispensación que separe el beneficio de una empresa de la salud de, esta vez sí, los pacientes y no de los clientes.
La versión genérica de un fármaco que ya está en el mercado, que puede suprimir y prevenir el VIH, seguiría produciendo un beneficio del 30% si se redujera el precio actual, afirman los investigadores.
Breve artículo que recoge un tema de una importancia y actualidad considerable, como es el tratamiento preexposición para el VIH Sida con Lanacapavir. De nuevo Gilead pone un precio abusivo de 42.250 $ a un fármaco que según estimaciones del nuestro bien conocido y estimado Andrew Hill costaría aproximadamente 40 $. En una situación en la que el objetivo para 2030, como señala Ouma, es impedir la aparición de nuevos casos de VIH/sida el resultado puede depender de disponer de medicamentos como éste. Obligar a Gilead a poner precios razonables es una obligación para frenar el desarrollo de la enfermedad. Si no, como afirma A. Hill, se deberían utilizar las licencias obligatorias.
Un nuevo medicamento descrito como «lo más cerca que hemos estado de una vacuna contra el VIH» podría costar 40 dólares (31 libras esterlinas) al año por cada paciente, mil veces menos que su precio actual, sugiere una nueva investigación.
Lenacapavir, vendido como Sunlenca por el gigante farmacéutico estadounidense Gilead, cuesta actualmente 42.250 dólares al año. Se insta a la empresa a que lo ponga a disposición en todo el mundo a mil veces menos que ese precio.
UNAids aseguró que podría «anunciar un avance en la prevención del VIH» si el medicamento estuviera disponible «rápidamente y asequible».
El lenacapavir administrado por inyección cada seis meses, puede prevenir la infección y suprimir el VIH en personas que ya están infectadas.
En un ensayo, en más de 5.000 mujeres en Sudáfrica y Uganda, el medicamento demostró un 100% de protección según los resultados anunciados por Gilead el mes pasado.
Lenacapavir actualmente tiene licencia para el tratamiento, no para la prevención.
En un estudio presentado el martes en la 25ª Conferencia Internacional sobre Sida en Múnich, los expertos calcularon que el precio mínimo para la producción en masa de una versión genérica, basado en los costos de los ingredientes y de la fabricación de lenacapavir, y permitiendo un 30% de beneficio, era de 40 dólares al año, asumiendo que 10 millones de personas lo usaran anualmente. A largo plazo, afirmaron que 60 millones de personas probablemente necesitarían tomar el medicamento de forma preventiva para reducir significativamente los niveles de VIH,.
El Dr. Andrew Hill, de la Universidad de Liverpool, que0 dirigió la investigación, comentó: «Tenemos una inyección que alguien podría recibir cada seis meses y no contraer el VIH. Eso es lo más cerca que hemos estado de una vacuna contra el VIH».
La mayor parte de la prevención del VIH actualmente se basa en píldoras diarias y medidas de barrera, como los condones.
Los activistas quieren que Gilead permita licencias genéricas, a través de Mrdicines Patent Pool respaldado por la ONU, en todos los países de bajos y medianos ingresos (LMIC), que representan el 95% de las infecciones por VIH. Mecanismos similares han estado en vigor en el mercado del tratamiento del VIH durante décadas, donde los países ricos pagan precios más altos que los más pobres.
Si eso no sucediera, dijo Hill, los países deberían considerar la emisión de licencias obligatorias que permitan la fabricación genérica frente a una emergencia de salud pública.
Gilead afirmó que era «demasiado pronto» para fijar el precio del lenacapavir para la prevención, ya que estaba a la espera de datos de ensayos clínicos y posibles presentaciones regulatorias, pero prometió «una estrategia para permitir una amplia, «Salvar vidas» al proporcionar una opción más discreta que las tabletas diarias para las personas que se enfrentaron al estigma debido a su estado de VIH o sexualidad.
Esto incluiría tanto «el suministro de Gilead en los países donde la necesidad sea mayor hasta que los empresas asociadas a través de licencias voluntarias puedan suministrar versiones de alta calidad y bajo costo de lenacapavir» y un programa de licencias voluntarias para «países de alta incidencia y recursos limitados». Gilead comenta que la elección de esos países estaba en curso.
Pero los activistas dijeron que era vital que todos los países de ingresos bajos y medios, incluidos los países de “ingresos medios altos” como Brasil, tuvieran acceso a formas genéricas de bajo costo del medicamento.
En el pasado, selecciones similares habían excluido a países donde la epidemia del VIH estaba creciendo más rápido, comentaron.
Los ensayos en los países de ingresos bajos y medios refuerzan aún más el argumento a favor del acceso universal, afirmó Hill, señalando como la Declaración de Helsinki sobre ética médica, dice que los ensayos solo deben realizarse en poblaciones que puedan beneficiarse de los resultados.
Joyce Ouma, oficial sénior de programas de Y+ Global, una red de jóvenes que viven con el VIH, dijo que un inyectable dos veces al año sería «transformador para los jóvenes como yo que viven o están en riesgo de VIH».
Ouma dijo: «No es exagerado decir que el objetivo de 2030 de poner fin a las nuevas transmisiones del VIH depende de Gilead para garantizar que las personas en el sur global tengan un acceso justo al lenacapavir».
Winnie Byanyima, directora ejecutiva de UNAids, afirmó que el tratamiento podría «salvar vidas» al proporcionar una opción más discreta que las tabletas diarias para las personas que enfrentaron estigmas debido a su estado de VIH o sexualidad.
Este artículo resultado de un proceso de investigación de Public Eye es de un notable interés. El autor ha analizado específicamente el caso de Roche y su fármaco Entresto. Nuestras lectoras y lectores pueden encontrar aquí una extraordinaria información sobre los procedimientos de “marañas de patentes” y “reverdecimiento de patentes” para prolongar el periodo de monopolio y conseguir así mantener los altos precios y conseguir y miles de millones de beneficio.
También el texto es muy ilustrativo de los procedimientos judiciales y de los acuerdos extrajudiciales con las empresas de genéricos para mantener el monopolio
Recomendamos vivamente su lectura
Las empresas farmacéuticas suizas a menudo se distinguen a nivel internacional por una acumulación de litigios legales destinados a evitar la competencia de los genéricos y mantener un precio alto en sus productos estrella. Sin embargo, esta estrategia pone en peligro el acceso a tratamientos asequibles. Es hora de actuar contra la multiplicación de patentes secundarias abusivas, sin valor añadido terapéutico y que solo sirven para llenar las arcas ya bien surtidas de Big Pharma. Una verdadera extorsión a costa de los seguros sociales, que Suiza debe denunciar en lugar de apoyar ciegamente.
Un nuevo medicamento no está protegido por una solo, sino por decenas, incluso a veces más de un centenar de patentes. Hablamos entonces de “maraña de patentes” (“patent thickets” en inglés). Estas se colocan a lo largo del tiempo, lo que significa que la duración del monopolio de un producto a menudo supera con creces los veinte años teóricos previstos por el Convenio sobre Propiedad Intelectual (ADPIC) de la Organización Mundial del Comercio (OMC). Una estrategia de acumulación de patentes sin fin calificada como “evergreening” en inglés.
Hay que distinguir dos tipos de patentes:
Las patentes primarias relativas a la molécula o moléculas y presentadas al principio de la fase de desarrollo.
Las patentes secundarias, presentadas justo antes o durante la fase de comercialización, que amplían el período de exclusividad comercial sin aportar un verdadero valor terapéutico.
Si bien cualquier patente es una excepción al libre mercado, las secundarias son las que tienen el mayor impacto en la competencia y los precios, especialmente porque han proliferado en los últimos años, especialmente en los Estados Unidos, donde se emiten más fácilmente.
Patentes secundarias concedidas en masa
Cada año, Suiza se jacta de ser uno de los “países más innovadores”, basándose simplemente en el número de patentes presentadas. Sin embargo, en el ámbito de los medicamentos, en cualquier caso, la gran mayoría es injustificada. De hecho, los grandes grupos farmacéuticos comprendieron rápidamente los beneficios financieros que podían obtener de estas murallas de patentes abusivas que bloquean el camino de sus competidores. En el otro extremo de la cadena, los pacientes tienen que pagar sus tratamientos a un precio alto durante más tiempo, sin ninguna justificación válida.
Recordemos que la patente es un derecho exclusivo que permite al titular de una invención prohibir que terceros la fabriquen y comercialicen. Pero es un derecho territorial: si una empresa farmacéutica quiere proteger su medicamento en varios países, debe solicitarlo en cada uno de ellos, excepto en Europa, donde la Oficina Europea de Patentes (OEB), que reúne a 39 países, incluida Suiza, tiene un procedimiento centralizado válido simultáneamente en todas estas jurisdicciones.
Recordemos también que una invención debe cumplir tres requisitos generales para ser patentada: (1) ser nueva (2) implicar una actividad inventiva (3) ser susceptible de aplicación industrial. Por lo tanto, una solicitud de patente sobre un medicamento no se juzga en función de la utilidad del tratamiento, sino que solo se tiene en cuenta el hecho de que sea un «nuevo invento», incluso si se trata solo de una modificación menor de un producto ya existente.
El acuerdo ADPIC deja un amplio margen de maniobra a los Estados miembros de la OMC para decidir qué invención merece una patente o no, siempre que se cumplan los tres requisitos. Así, según la legislación vigente y la minuciosidad con la que se examinan las solicitudes, las patentes se conceden en masa (como en los Estados Unidos); de forma un poco más restringida porque a veces se impugna (como en Europa); o con moderación debido a cláusulas más restrictivas destinadas a evitar recompensar pseudoinnovaciones que ponen en peligro el derecho a la salud (como en la India). Estos enfoques tienen consecuencias muy diferentes en términos de competencia y acceso a los medicamentos, con una llegada al mercado más o menos tardía, según el país, de genéricos vendidos a precios más bajos.
Estados Unidos, auténtico “El Dorado” de la farmacia
Como en muchos otros sectores, Estados Unidos marca el tono en el campo farmacéutico. Con más de 600 mil millones de dólares estadounidenses anuales, el país del Tío Sam representa por sí solo más de la mitad del mercado farmacéutico mundial. Un campo de juego esencial para Roche y Novartis, respectivamente segundo y octavo del mundo en términos de facturación en 2023.
Los gigantes de Basilea son miembros desde hace mucho tiempo del poderoso lobby farmacéutico en los Estados Unidos (Pharmaceutical Research and Manufacturers of America o PhRMA), que está en la calle del Congreso y la Casa Blanca. El CEO de Novartis incluso asume su presidencia desde 2023. En los Estados Unidos, las empresas farmacéuticas se benefician de numerosos incentivos y grandes ventajas fiscales en el ámbito de la investigación, así como de una política de patentes muy generosa y de un sistema judicial propicio para iniciar litigios a toda marcha. El procedimiento de autorización de comercialización también está estrechamente relacionado con el estatuto de las patentes, lo que no es el caso en Europa. Y, la guinda del pastel: hasta la fecha no existe un verdadero control estatal de los precios.
Por lo tanto, los grandes grupos buscan lanzar primero sus nuevos productos en los Estados Unidos, para poder proteger su invención el mayor tiempo posible (a veces durante 40-50 años) y obtener un precio muy alto en el mercado estadounidense, que luego utilizarán como base de negociación en otros países, por ejemplo en Europa, donde el control de precios es un poco más estricto.
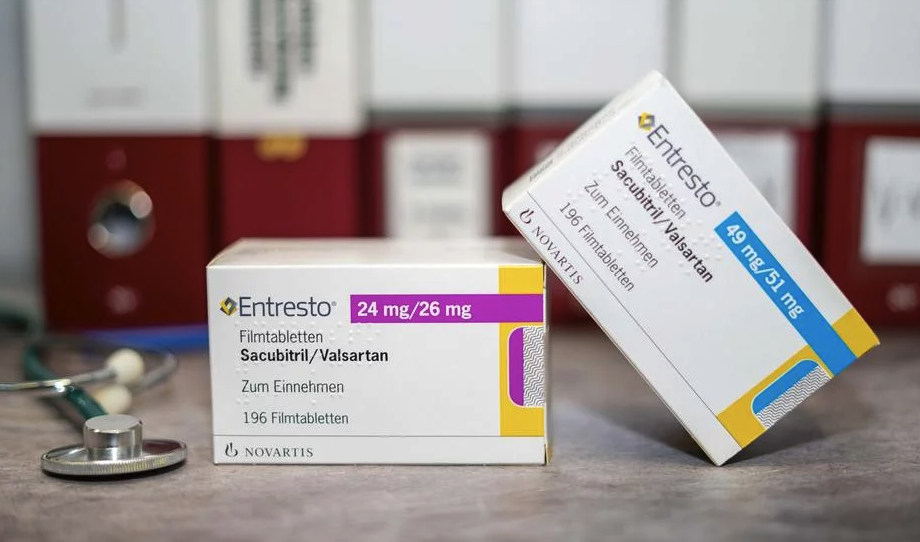
El Entresto, punta de lanza de Novartis
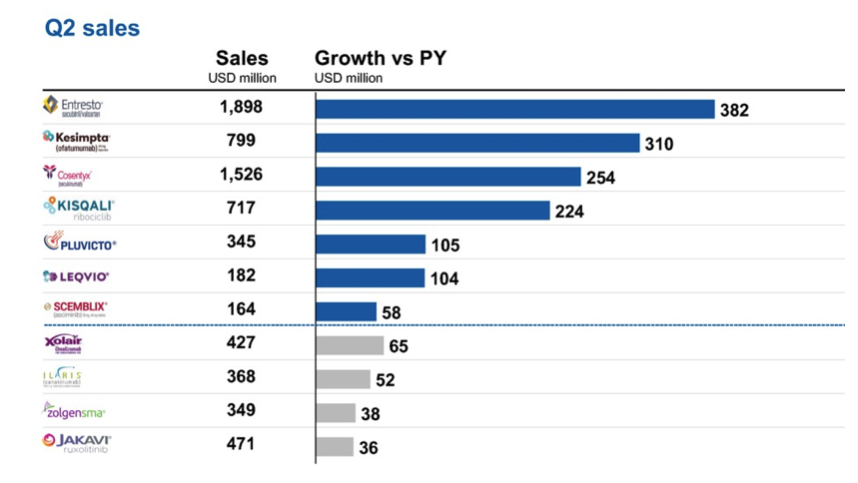
Para ilustrar cómo la farmacia explota su posición de fuerza para evitar cualquier competencia, hemos estudiado el caso del Entresto de Novartis. Después de un comienzo bastante lento, este tratamiento para la insuficiencia cardíaca, lanzado en julio de 2015 en los Estados Unidos y poco después en Suiza y Europa, vio explotar sus ventas en 2021, gracias a la obtención de una extensión de indicación para diferentes tipos de insuficiencia cardíaca. En Suiza, sus ventas anuales se han más que duplicado, pasando de 18 a más de 39 millones de francos entre 2019 y 2023, según la caja de salud Helsana. En 2023, Entresto generó los mayores ingresos del grupo a nivel mundial, con más de 6 mil millones de dólares (aproximadamente el 13% de las ventas totales). En apenas ocho años, Novartis ya ha recaudado más de 20 mil millones de dólares en ventas gracias a este producto.
Su precio oficial en Suiza para un mes de tratamiento es de unos 130 francos (2,3 francos por comprimido). Como suele ser el caso de los medicamentos, es cuatro veces más alto en los Estados Unidos: 668 dólares al mes, mientras que es un poco más barato en la India (10 200 rupias, o unos 103 francos al mes). Este precio puede parecer irrisorio en comparación con los contra el cáncer, pero el margen sigue siendo importante debido a una fuerte demanda y un coste de producción extremadamente bajo: 0,13 francos por comprimido.
Además, Entresto es una combinación de dos moléculas antiguas, incluido el valsartán, que ha hecho los mejores días de Novartis como tratamiento para la hipertensión en los últimos 25 años bajo el nombre de marca Diovan, con más de 65 mil millones de dólares en ingresos hasta la fecha. Desde un punto de vista comercial, el Entresto es, por tanto, un intento de Novartis de prolongar las enormes ventas de su predecesor Diovan, al tiempo que amplía su público objetivo a los pacientes con insuficiencia cardíaca. En resumen: el premio gordo
La inversión de Novartis para desarrollar el Entresto se ha amortizado desde hace mucho tiempo, con un margen de beneficio astronómico. Sin embargo, el gigante de Basilea siempre quiere más y ha iniciado, en 2019, una verdadera saga judicial en Estados Unidos e India para retrasar al máximo la llegada al mercado de competidores genéricos. Aquí es donde entran en juego las patentes secundarias.
Una jungla de patentes “frívolas”
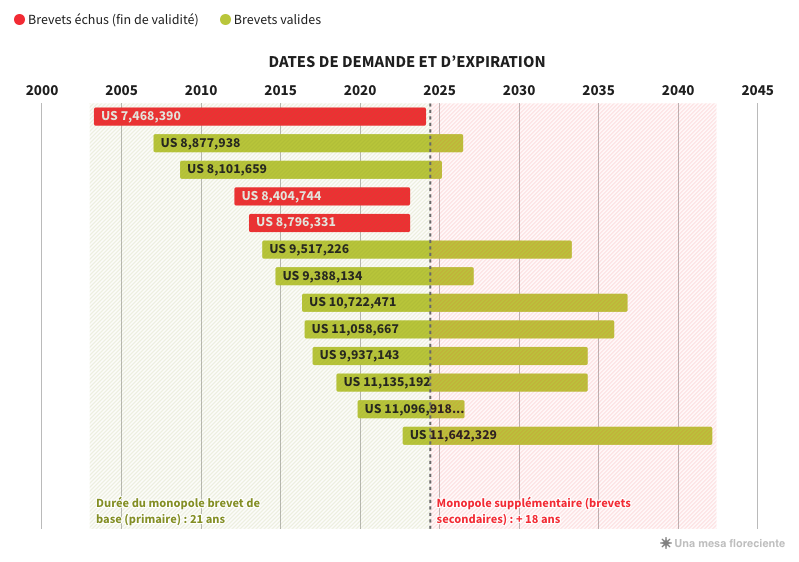
Novartis ha obtenido al menos trece patentes sobre este producto en los Estados Unidos, lo que teóricamente le otorga una exclusividad comercial de casi 40 años, el doble del estándar previsto por las normas de la OMC (véase la tabla ). Más allá del número, es el tipo de patentes y sus fechas de presentación lo que llama la atención. El medicamento, una combinación de dos moléculas, se ha mantenido igual desde el principio. Solo han cambiado su uso (indicación), su dosis y otros aspectos como su método de administración. Sin embargo, cada vez se han presentado y concedido nuevas patentes secundarias. ¿Para qué ganancias terapéuticas? Prácticamente ninguna. Por otro lado, la duración del monopolio se ha ampliado 18 años, hasta 2042.
PATENTES DE ESTADOS UNIDOS (ENTRESTO, NOVARTIS)
Con la patente primaria vencida, el período de monopolio de 20 años previsto por las normas de la OMC debería terminar. Pero con todas las demás patentes secundarias obtenidas, Novartis puede teóricamente mantener los genéricos fuera durante 18 años más suplementarios en los EE.UU
En Europa, hay al menos nueve solicitudes de patentes, tres de las cuales están siendo examinadas por la OEP. Solo tres patentes son válidas hasta la fecha (véase la tabla ). La patente primaria del Entresto ha expirado desde 2023, pero se beneficia de una extensión de protección en Suiza hasta enero de 2028, concedida por las autoridades suizas. La protección del Entresto en Europa, todas las patentes concedidas combinadas, se extiende teóricamente hasta mayo de 2036, pero si las tres solicitudes en curso tienen éxito, será de 40 años, es decir, el doble del estándar de la OMC. Se han revocado dos patentes secundarias (una por el titular; la otra por oposición), lo que demuestra que no se habrían concedido si el examen por parte de la OE hubiera sido más minucioso.
PATENTES EUROPEAS Y EXTENSIONES SUIZAS (ENTRESTO, NOVARTIS)
En Europa también la patente primaria ha caducado desde 2023, pero gracias a otros privilegios nacionales su protección durará hasta 2028 en Suiza. Gracias a las patentes secundarias obtenidas y futuras (tres solicitudes en curso), Novartis podría impedir la llegada de genéricos de Entresto hasta 2043, es decir, 40 años de monopolio en total
El panorama sigue siendo diferente en la India, donde se han concedido cinco patentes sobre el Entresto (comercializado bajo la marca Vymada), cuatro de ellas secundarias (véase la tabla siguiente). La patente primaria (que caducó en enero de 2023) fue impugnada en los tribunales en 2019 por cuatro fabricantes de genéricos, en vano. La segunda patente, presentada en 2006, fue objeto de nueve oposiciones antes de su concesión, según lo autorizado por la ley india, pero finalmente se concedió. Entonces se presentaron nuevos recursos; el procedimiento sigue en curso. En cuanto a las otras tres patentes, bien podrían ser impugnadas posteriormente en los tribunales por empresas indias. Los desafíos: no retrasar la comercialización de equivalentes genéricos más asequibles (al menos un 50% más baratos que el original) y mejorar el acceso a este producto, en un país donde la mayoría de los pacientes pagan los tratamientos médicos de su bolsillo.
Con sus patentes primarias vencidas y más de 85 mil millones de dólares recaudados en 25 años, gracias a Diovan y Entresto, es hora de que Novartis finalmente deje espacio para los competidores. Pero al gigante de Basilea no le importa y sigue acudiendo sistemáticamente a la justicia para obstaculizar a sus competidores a través de patentes secundarias abusivas.
PATENTES INDIA VYMADA (ENTRESTO, NOVARTIS)
La legislación india es más estricta en términos de concesión de patentes, con cláusulas de salud pública para luchar contra las patentes secundarias abusivas, como lo permiten los acuerdos de la OMC. Sin embargo, si todas las oposiciones en curso o futuras fracasan, Novartis podría beneficiarse de un monopolio total de 33 años, mucho más allá de los estándares de la OMC.
Quejas en ráfaga en los Estados Unidos
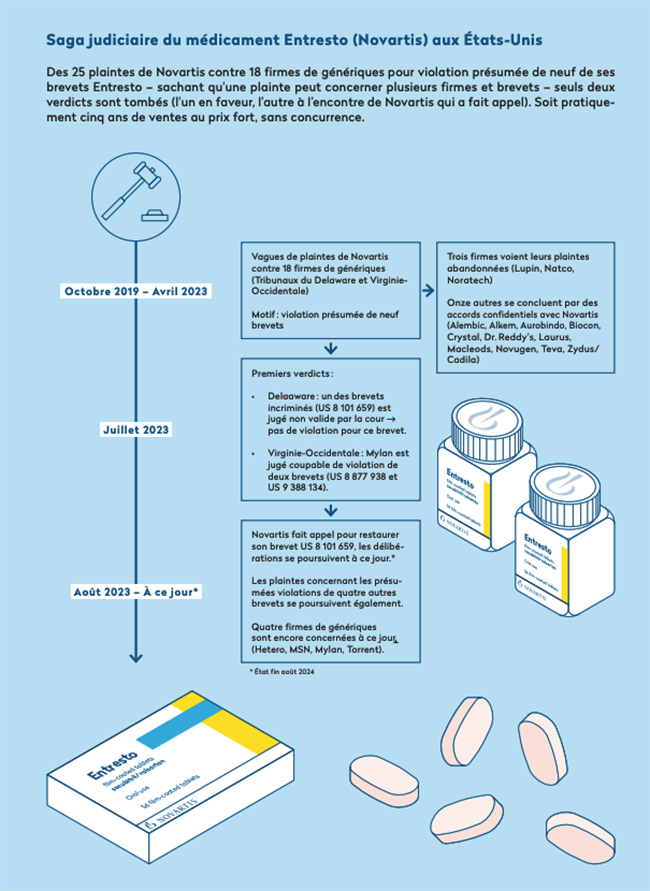
Nuestra exhaustiva investigación ha permitido rastrear las numerosas denuncias presentadas por Novartis en los Estados Unidos por supuesta infracción de patentes con Entresto, sobre la base de documentos judiciales a los que hemos tenido acceso (ver la cronología siguiente).
En el inicio de esta investigación, una observación sorprendente: durante la última década, las empresas farmacéuticas suizas han acudido a la justicia de forma casi rutinaria en los Estados Unidos o en la India con el objetivo de evitar -o al menos retrasar fuertemente- la competencia, ya sea para Entresto o Gilenya (contra la esclerosis múltiple) sobre Novartis, o para Esbriet (contra la fibrosis pulmonar) o sus tratamientos contra el cáncer de mama (Herceptin en el pasado, actualmente Perjeta) de Roche. Hemos estudiado cada uno de estos casos, pero nos centraremos aquí en el emblemático del Entresto.
En los Estados Unidos, Novartis presentó, entre octubre de 2019 y octubre de 2022, no menos de 25 denuncias por presunta infracción de nueve de sus patentes de Entresto contra 18 empresas farmacéuticas que han informado de su intención de comercializar versiones genéricas allí. Hay que tener en cuenta que todas estas quejas, antes de la comercialización, eran puramente preventivas. Las empresas en cuestión no vendían entonces ningún genérico de Entresto en el mercado estadounidense, sino que simplemente habían iniciado el largo procedimiento de aprobación con la Administración de Alimentos y Medicamentos (FDA), para estar listos cuando finalizara la exclusividad comercial. Estos enjuiciamientos preventivos son una particularidad de la ley estadounidense, llamada «enlace de patentes» en inglés, que vincula el estado de las patentes al procedimiento de autorización de comercialización. Una situación que, afortunadamente, Europa no conoce. El papel de una agencia de medicamentos como Swissmedic es asegurarse de que los tratamientos a homologar sean seguros y eficaces, no jugar al gendarme de la competencia.
De las 18 empresas demandadas por Novartis, tres vieron la denuncia en su contra abandonada, por falta de violación constatada. Otros once han llegado a un acuerdo confidencial con Novartis, probablemente comprometiéndose a no comercializar sus genéricos antes de una fecha acordada entre las partes, a cambio del cese del proceso.
Este tipo de acuerdo adopta, en general, dos formas en el ámbito farmacéutico:
La concesión de una licencia contra el pago de regalías, válida a partir de una fecha determinada;
El “pay-for-delay”, una táctica clásica, especialmente cuando una patente se tambalea. El fabricante de un medicamento original paga entonces una cantidad determinada a una empresa competidora para animarla a posponer el lanzamiento de su genérico. Esta práctica, también frecuente en Europa, ha sido fuertemente criticada en varias ocasiones por las autoridades de competencia. También cuesta mucho a los sistemas de salud, ya que el medicamento sigue vendiéndose a alto precio hasta la llegada de un genérico. En el caso del Entresto, parece que se han celebrado acuerdos de licencia, aunque no es posible ninguna certeza, ya que los documentos judiciales están sellados y ocultados .
La cronología de las denuncias presentadas por Novartis en los Estados Unidos por presunta infracción de patentes en Entresto se descarga aquí en PDF.
En julio de 2023 se emitieron los dos únicos veredictos hasta la fecha en esta saga judicial:
En un caso, después de cuatro años de procedimiento con grandes refuerzos de expertos y abogados, una de las nueve patentes de Entresto en cuestión fue invalidada por el tribunal de Delaware. Novartis recurrió inmediatamente al nivel federal (el procedimiento está en curso).
En el otro veredicto, el tribunal de Virginia Occidental se pronunció a favor del gigante de Basilea, confirmándose la violación de dos patentes contra la empresa Mylan. Este último no apeló, y no se puede detectar ninguna otra información a partir de los documentos judiciales, pero podemos imaginar que se ha llegado a un acuerdo confidencial. Hay que tener en cuenta en que esto se jugó a media molécula de agua en la fórmula química entre el producto original de Novartis y el genérico de Mylan para inclinar el veredicto en una dirección u otra, según la decisión del tribunal. Esto demuestra la complejidad del procedimiento, pero también el considerable incremento de tiempo que pueden obtener los gigantes farmacéuticos gracias a tales denuncias judiciales.
Actualmente, las quejas de Novartis se refieren “solo” a cuatro empresas y a una supuesta infracción de cuatro patentes (cinco si Novartis gana su apelación). No se sabe cuándo se emitirán los próximos veredictos, pero el procedimiento aún podría durar.
Entre mayo y agosto de 2024, siete versiones genéricas del Entresto finalmente obtuvieron luz verde de la FDA, pero esta autorización aún no significa que pronto puedan comercializarse y acceder a los pacientes. Porque Novartis vuelve a acudir a la justicia, con una demanda civil presentada el 30 de julio, esta vez contra la FDA por violación de sus procedimientos de homologación (Novartis Pharms Corp. v. Xavier Becerra y Robert Califf, Caso No. 1:24-cv-02234, US District Court of Columbia, 30/07/2024). Aunque el tribunal desestimó en primera instancia la moción de Novartis para bloquear la autorización de la FDA, estas empresas podrían ver que la comercialización de sus genéricos de Entresto se retrase aún más en los Estados Unidos, dependiendo del resultado de esta queja, así como de otros litigios de patentes en curso.
Mientras tanto, Novartis puede seguir ganando miles de millones de dólares adicionales gracias a sus patentes secundarias «frívolas», un ejemplo perfecto de evergreening. Todo esto a costa de los pacientes y de los seguros sociales, una verdadera estafa.
Una saga judicial también en la India
Dado que la India siempre se negó a introducir un sistema que vinculara el estado de las patentes con el procedimiento de homologación («enlace de patentes»), las versiones genéricas de Vymada (marca comercial de Entresto en la India) obtuvieron una autorización de comercialización en 2019. Las perspectivas son realmente jugosas, con un mercado de cardiología estimado en 2.500 millones de francos y más de 650 000 nuevos casos de insuficiencia cardíaca diagnosticados cada año. En 2019, Novartis demandó a cuatro fabricantes de genéricos indios afectados, que luego contraatacaron solicitando la revocación de la patente primaria (IN 229051). El Tribunal Superior de Delhi finalmente dio la razón al gigante de Basilea en 2021, prohibiendo a las empresas locales fabricar y comercializar sus versiones genéricas, al menos hasta que expire la patente primaria (enero de 2023).
A continuación, se prestó atención a la segunda patente, secundaria (IN 414518), que se había concedido en la India a pesar de nueve oposiciones debidamente argumentadas, y que extendió la exclusividad comercial de Novartis hasta noviembre de 2026. Por lo tanto, varias empresas de genéricos han recurrido a la justicia, desde 2022, para intentar revocar esta patente secundaria después de su concesión. En un primer momento, el Tribunal Superior de Delhi suspendió, en enero de 2023, la patente secundaria en cuestión, antes de revocar su decisión unos días después, confirmando su validez. El resto es aún más confuso, entre el contraataque de las empresas genéricas para intentar revocar la patente secundaria y las apelaciones de Novartis, que sepamos, sin veredicto hasta la fecha. Y la cuestión de las otras tres patentes secundarias, con una duración de protección teórica hasta febrero de 2037, tampoco está resuelta.
Aunque este no ha sido el caso hasta ahora para Entresto, la farmacéutica suiza se ha roto los dientes varias veces en el pasado en la India con las oposiciones a las patentes previas a la concesión (o “pre-grant”). Empezando por el emblemático caso del anticancerígeno Glivec de Novartis, al que las autoridades indias le habían negado su patente primaria. Solo unos pocos países, como India o Tailandia, utilizan esta flexibilidad legal consagrada en los acuerdos de la OMC. Europa (a excepción de Portugal) y Estados Unidos no prevén estos procedimientos previos a la concesión, que tienen el don de irritar a Big Pharma, que los considera un obstáculo para el buen funcionamiento de sus negocios. En su acuerdo bilateral de libre comercio celebrado recientemente con la India, Suiza también ha logrado debilitar estas posibilidades de intervención previa. Muy malas noticias para el acceso a los medicamentos y la salud pública.
Novartis demanda a la administración Biden
En los Estados Unidos, el caso Entresto no se limita a un tira y afloja entre las empresas farmacéuticas. Novartis también presionó directamente a la FDA para que el policía estadounidense de medicamentos no homologara ningún genérico de su producto durante su período de exclusividad comercial, siempre buscando conseguir tiempo adicional.
En septiembre de 2021, el Departamento de Justicia anunció la apertura de una investigación civil por posibles remuneraciones pagadas a la profesión médica con el fin de impulsar las ventas de Entresto. Novartis ya ha estado en el punto de mira de las autoridades estadounidenses por prácticas comerciales ilegales. En 2020, la empresa de Basilea tuvo que pagar una multa de más de 670 millones de dólares para resolver un caso de corrupción relacionado con varios de sus productos (incluido Diovan, predecesor de Entresto). Desde entonces, no se ha hecho pública ninguna otra comunicación sobre la última investigación sobre Entresto.
En agosto de 2022, es el problema de la difusión: Novartis es demandada por las universidades de Michigan y Florida del Sur por una posible violación de su patente que cubre una técnica de fabricación utilizada para el Entresto. Se desconoce el resultado de este caso, pero podría haberse resuelto en forma de compensación financiera de Novartis a ambas universidades.
Por último, gracias a la aprobación de la Ley de Reducción de la Inflación (Inflation Reduction Act o IRA), obtenida en agosto de 2022 por la administración Biden frente al poderoso lobby farmacéutico PhRMA, el seguro público Medicare (para personas mayores de 65 años) ha obtenido, por primera vez en su historia, la posibilidad de negociar directamente el precio de los tratamientos más caros en términos de cobertura. Un año después, se hizo pública la lista de los primeros diez medicamentos que debían pasar por este nuevo procedimiento, con un nuevo precio regulado aplicable a partir de 2026.
Entre estos productos prioritarios: el Entresto de Novartis, le costó a Medicare alrededor de 2.900 millones de dólares en 2023 para unos 600.000 asegurados. Medicare aspiraba a reducir su precio en al menos un 25%. La reacción no se hizo esperar: Novartis demandó al gobierno de Estados Unidos el 1 de septiembre de 2023, calificando esta reforma de inconstitucional. La empresa de Basilea considera que es similar a una “expropiación de bienes privados” y que corre el riesgo de “poner en peligro la investigación de medicamentos innovadores”.
Todas las grandes farmacéuticas afectadas, así como su organización paraguas, también han acudido a la justicia, gritando que viene el lobo. Incluso Roche, cuyo nombre no aparece en esta primera selección, fue con un chantaje, amenazando con retrasar la comercialización de nuevos productos vitales debido a esta reforma. Como a menudo, Big Pharma se une para evitar cualquier precedente desafortunado que pueda ir en contra de su modelo de negocio, especialmente en su Eldorado en los Estados Unidos, donde las empresas eran hasta ahora todopoderosas en la fijación de precios.
Sin embargo, una investigación de la ONG Public Citizen mostró que en 2022, las empresas farmacéuticas afectadas por estas negociaciones invirtieron en la recompra de acciones, el pago de dividendos a los accionistas y el salario de sus directivos en un promedio de 10 mil millones de dólares más que en investigación y desarrollo (I+D); para Novartis, fueron 18 mil millones frente a 10 mil millones para I+D. Suficiente para relativizar considerablemente su amenaza con respecto a la innovación.
Sin embargo, Novartis se ha resignado a entrar en negociaciones a pesar de su denuncia en curso. La razón: impuestos de hasta el 95% de la facturación del producto en cuestión si no lo hiciera. También pudo presentar una contraoferta al precio ofrecido por Medicare. A mediados de agosto de 2024, Medicare hizo públicos los nuevos precios negociados. Estos mostraron una reducción de más del 50% en el precio del Entresto (295 dólares), que Novartis criticó de inmediato. Si bien otras dos quejas de Big Pharma ya han sido rechazadas, la de Novartis contra el gobierno de Biden sigue pendiente.
Suiza debe actuar contra las patentes abusivas
El “evergreening”, o la acumulación abusiva de patentes secundarias sobre productos terapéuticos, representa un freno al acceso a los medicamentos, así como un enorme coste adicional para los pacientes y la sociedad. En Suiza, los medicamentos representan 1 franco de cada 4 de los gastos del seguro médico obligatorio, de los cuales el 75% se debe a productos patentados, según un análisis del Consejo Federal. ¿Con qué cantidad de patentes «frívolas» que permiten mantener un monopolio -y el alto precio que conlleva- mucho más tiempo que la duración prevista por las normas de la OMC? Imposible de cuantificar, a falta de estudios precisos sobre el tema en Europa. Sin embargo, se puede apostar a que esta proporción es alta si se compara el número limitado de nuevos medicamentos lanzados cada año en el mercado con todas las patentes farmacéuticas presentadas.
Según la ONG I-MAK, el abuso de patentes de los diez medicamentos más vendidos en Estados Unidos asciende a decenas de miles de millones de dólares en costes adicionales para el sistema de salud cada año. El gobierno de Estados Unidos finalmente levanta el tono contra estas selvas de patentes que alimentan la codicia de Big Pharma, y está considerando reformas. ¿Está el viento girando al otro lado del Atlántico?
Suiza, por su parte, se niega sistemáticamente a actuar contra los abusos de propiedad intelectual relacionados con el acceso a los medicamentos en los foros multilaterales, como se ha visto durante la crisis de Covid (en la OMC) y actualmente en el marco del Tratado Internacional sobre Pandemias, en negociación en la Organización Mundial de la Salud (OMS). Peor aún, las autoridades suizas buscan reforzar aún más la propiedad intelectual o, si no lo logran, limitar el margen de maniobra de los países de bajos ingresos para luchar contra los abusos, como se ha visto en el marco del acuerdo bilateral de libre comercio celebrado en marzo con la India.
Como miembro de la OEP, que expide las patentes europeas de la industria farmacéutica, Suiza podría actuar a este nivel para ofrecer un examen más detallado de las solicitudes. Aunque Europa concede menos que Estados Unidos, todavía se emiten demasiadas patentes no merecidas, como lo ilustra nuestra oposición de 2019 sobre el anticancerígeno Kymriah, tras la cual Novartis revocó la patente impugnada antes de cualquier debate contradictorio. Es mejor evitar que se expidan patentes abusivas, en lugar de tener que luchar contra ellas después en litigios largos y costosos. Para ello, es imperativo definir normas de patentabilidad más estrictas y aplicarlas.
Suiza se había opuesto durante mucho tiempo a las patentes de medicamentos, considerando que se trataba de un bien esencial como ningún otro, antes de cambiar radicalmente de posición. Sin llegar a tal cambio, ¿por qué no empezar por luchar contra las prácticas abusivas de sus farmacias, que tienen consecuencias perjudiciales para la salud y las finanzas públicas, tanto en Suiza como en otros lugares?
La Federación de Asociaciones para la Defensa de la Sanidad Pública (FADSP) se ha distinguido durante años por su constante y valiosa defensa de una sanidad pública de calidad. En este artículo el autor recoge el planteamiento claro y decidido de la FADSP para modificar el actual sistema de patentes de fármacos mediante una reflexión de la reciente sentencia sobre Apixabans.
LaFederación de Asociaciones para la Defensa de la Sanidad Pública (FADSP) considera que el sistema de patentes hay que cambiarlo y premiar la innovación con sistemas más alineados con los intereses públicos.
Si la salud es un derecho humano fundamental, indispensable para el ejercicio de los demás derechos humanos, el acceso a los medicamentos es parte de ese derecho. El precio excesivo de los medicamentos (muy por encima del coste de fabricación y de I+D), además de suponer una barrera importante al acceso, provoca que la calidad de otras prestaciones se vea mermada y que se ponga en riesgo la estabilidad del sistema sanitario público.
Al otorgar las patentes, los Gobiernos conceden a las empresas un periodo de tiempo de exclusividad en el mercado como supuesta forma de mantener y mejorar la innovación. Durante esos años, el propietario de la patente puede fijar un precio del fármaco muy por encima de los costes, puede recuperar los costos de su inversión en I+D y obtener un beneficio supuestamente adecuado al riesgo que soporta. Pero en la práctica el sistema de patentes permite que los laboratorios impongan precios abusivos a los sistemas públicos de salud.
La Federación de Asociaciones para la Defensa de la Sanidad Pública (FADSP) aboga por cambiar el actual sistema de patentes de fármacos. Estas son responsables de la hegemonía de la financiación privada, el monopolio comercial y las estrategias de extensión de patentes, la investigación en cerrado, el marketing como instrumento de ventas y la necesidad de retornos en el corto plazo (que favorecen estrategias especulativas, agresivas conspiraciones y acuerdos en la sombra y una elevada prevalencia de delitos corporativos).
La FADSP considera que el sistema de patentes es contrario a la sostenibilidad, la cooperación y la ciencia independiente. Hay que cambiarlo y premiar la innovación con sistemas más alineados con los intereses públicos, para ello se precisa impulsar reformas legislativas y estructurales orientadas a promover licencias abiertas, compartidas y no exclusivas, donde la propiedad intelectual no suponga una barrera en el acceso a los tratamientos que pudieran finalmente desarrollarse. También impulsar plataformas públicas para investigación clínica que potencien la financiación de investigación básica y de Ensayos Clínicos No Comerciales que aglutine los estudios independientes y generen derechos y patentes de titularidad pública.
Puede ser muy clarificador analizar el más reciente caso de “guerra de patentes” ocurrido respecto al apixabán, principio activo indicado para el tratamiento de diversas situaciones proclives a la formación de trombos. El certificado complementario de protección de la patente (que propiamente dicha ya expiró) del medicamento Eliquis de la farmacéutica Bristol Myers Squibb (BMS), que ha comercializado el apixabán, vence en noviembre de 2026, pero el laboratorio Teva Pharma pleiteó en los tribunales utilizando el argumento de plausibilidad, una vía abierta el año pasado ante la Oficina Europea de Patentes (EPO) para producir el primer genérico de apixabán: Apixabán Tevagen del laboratorio Teva Pharma comercializado con un precio casi un 50% inferior al de Eliquis.
Un principio fundamental del derecho de patentes es que el alcance del monopolio de la patente debe estar justificado por la contribución técnica del titular de la patente a la técnica. Al considerar la actividad inventiva, es necesario considerar qué problema técnico resuelve la invención reivindicada; si no es plausible que la invención resuelva ningún problema técnico, entonces el titular de la patente no ha hecho ninguna contribución técnica y la invención no implica una actividad inventiva.
El Juzgado de lo Mercantil 4 de Barcelona dio en enero la razón a Teva, y cuando la farmacéutica tenía listo el lanzamiento del genérico para el 1 de marzo, BMS recurrió a otro juzgado de Madrid y logró que este lo paralizara de forma cautelar. La situación creó un conflicto de jurisdicciones que tardó cerca de dos meses en ser resuelto por el Supremo, en este caso a favor de Teva (en el sentido de conceder la jurisdicción al juzgado de Barcelona). Dos datos ilustran las enormes cifras de negocio que dependen de estos procesos. El primero es que el retraso de dos meses en la salida al mercado del genérico por una medida cautelar hizo que la sanidad pública no pudiera ahorrar en primavera 23 millones de euros. El segundo, que en los cuatro meses que el genérico ha estado en el mercado desde mayo, las comunidades autónomas han ahorrado más de 45 millones de euros.
Ahora, una sentencia dictada el pasado 18 de julio por la Audiencia de Barcelona da la razón a BMS en sus argumentos de que la protección del Eliquis debe seguir vigente, devuelve la partida a la casilla de salida, aunque de forma provisional. Teva tiene previsto presentar un recurso ante el Tribunal Supremo, aunque este podría tardar más de un año en resolverse. Los litigios entre farmacéuticas suelen incluir reparaciones entre ellas por las que las compañías que pierden un caso indemnizan a las que lo ganan, pero esto no ocurre con la sanidad pública. Es decir, si el Supremo diera de aquí a un año la razón a Teva, las comunidades autónomas no serían resarcidas por los 138 millones adicionales (380.000 euros diarios) que pagarán durante los próximos 12 meses.
Divulgación de información sobre las actividades de lobby en EE. UU: Cómo las grandes farmacéuticas lucharon contra la exención de los ADPIC y presionan para lograr una protección sólida de la propiedad intelectual en el Acuerdo sobre la Pandemia
OTRAS FUENTES. Revista Nº 33 Octubre 2024.
Peter Maybarduk & Michael Weinstein.
Peter Maybarduk, Director del Grupo de Acceso a Medicamentos de Public Citizen ( pmaybarduk@citizen.org ); Michael Weinstein, Presidente de la AIDS Healthcare Foundation (Contacto de prensa: Guilherme Faviero, AHF Global Public Health Institute, Guilherme.faviero@ahf.org ), en Priti Patnaik en Génova Health Files, 28-10-2024, https://substack.com/home/post/p-150770965
Recogemos aquí un texto seleccionado por Priti Patnaik en Génova Health Files de una investigación realizada por Public Citizen sobre las acciones de lobby de la industria farmacéutica en su esfuerzo para mantener fuertes protecciones de la propiedad intelectual y las implicaciones que han supuesto y suponen para la salud pública durante y después de COVID-19.
La información destaca las acciones de lobby en el marco de las discusiones para abordar decisiones políticas en las negociaciones sobre la exención de los ADPIC en la OMC y de la negociación actualmente en desarrollo el sobre el Acuerdo sobre la Pandemia en la OMS.
Los hallazgos del estudio muestran que las corporaciones, los grupos comerciales y otras organizaciones desplegaron a más de 500 lobistas para persuadir a los legisladores y los responsables de las políticas estadounidenses durante la COVID-19 para que moldearan la posición global del gobierno de Estados Unidos sobre la propiedad intelectual, y casi el 90 por ciento de estos lobistas se oponían a la exención de los ADPIC. Según Public Citizen, en 2022, el año en el que se contrataron más lobistas, las entidades opuestas a la exención de los ADPIC superaron en número a las contratadas por los partidarios en una proporción de 32 a 1.
Veinte entidades revelaron que habían ejercido presión sobre la exención durante la primera mitad de 2024, cuando concluyeron las negociaciones sobre la exención de los ADPIC. Estas entidades son en su mayoría empresas farmacéuticas o de biotecnología y sus asociaciones comerciales relacionadas.
Estas revelaciones son cruciales a la luz no solo de las negociaciones en la OMC, sino también de las negociaciones en curso hacia un Acuerdo sobre la Pandemia en la OMS, donde los países desarrollados han luchado por una mayor protección de la propiedad intelectual en el contexto de futuras emergencias.
Durante la pandemia de COVID-19, las 20 compañías farmacéuticas más grandes del mundo gastaron casi tanto en enriquecer a los accionistas como en investigación y desarrollo.
Los gigantes de la industria Amgen y Novo Nordisk pagaron dividendos a los accionistas que duplicaron su gasto en I+D, mientras que Novartis y AbbVie dirigieron más de 10 mil millones de dólares más a los accionistas que a la investigación.
A medida que el virus barría el mundo, exponiendo profundas desigualdades en el acceso a las medidas contra la pandemia, las naciones del Sur Global lucharon por asegurar vacunas y tratamientos que salvaban vidas, ya que los países de altos ingresos acaparaban suministros, imponían controles de exportación y compraban la mayoría de las existencias disponibles. Con el tiempo, aprenderíamos que se estima que solo el acaparamiento de vacunas se ha cobrado más de 1,3 millones de vidas, con un impacto desproporcionado en los países de bajos y medianos ingresos (LMIC).
A pesar de los pagos sustanciales de los accionistas que excedieron muchos de los presupuestos de I+D, el lobby farmacéutico estaba trabajando arduamente para socavar las propuestas globales destinadas a promover un mayor acceso a vacunas, terapias y diagnósticos en el Sur Global. Armada con la impugnada premisa de que tales esfuerzos «sofocarían la innovación», la industria presionó ampliamente contra la aplicación de la exención de los aspectos relacionados con el comercio de los derechos de propiedad intelectual (TRIPS) que habría suspendido temporalmente las restricciones de propiedad intelectual de la Organización Mundial del Comercio (OMC) en productos que salvan vidas.
La exención, propuesta originalmente por India y Sudáfrica en octubre de 2020, buscaba suspender temporalmente disposiciones específicas del acuerdo sobre los ADPIC de la OMC para garantizar que las normas de propiedad intelectual de la OMC no crearan barreras para el acceso oportuno a las contramedidas para combatir la pandemia. Esta propuesta, de ser adoptada, habría apoyado los esfuerzos para ampliar la producción y abordar las brechas mundiales en el suministro de vacunas, diagnósticos y tratamientos hasta que la vacunación generalizada estuviera disponible. Si bien la exención por sí sola no habría sido una solución completa al problema del acceso, habría hecho una contribución significativa al eliminar las barreras comerciales a las contramedidas críticas durante la peor emergencia sanitaria en más de cien años. También habría señalado una cooperación mundial esencial en la lucha contra el acaparamiento de productos sanitarios relacionados con la pandemia y lo que se ha denominado el apartheid de la vacuna.
A pesar del apoyo de casi 100 naciones en desarrollo, y más de 170 ex jefes de gobierno y premios Nobel, la exención se enfrentó a una oposición significativa de algunas naciones ricas. Sujetos a los intensos esfuerzos de lobby por parte de la industria farmacéutica, muchos países de altos ingresos, incluidos el Reino Unido, los miembros de la UE e inicialmente los Estados Unidos, trataron de retrasar la propuesta a través de tácticas de demora que se extendieron durante unos asombrosos 18 meses, hasta que la exención estuvo muerta definitivamente. Todo esto ocurrió durante un período crítico de la peor crisis sanitaria mundial en la memoria reciente, cuando una acción rápida en todos los frentes hubiera sido vital para abordar las desigualdades y salvar vidas.
En Europa, donde una parte sustancial de las economías de muchas naciones proviene de la industria farmacéutica, el Observatorio de Europa Corporativa reveló que, a partir de 2021, más de 40 empresas farmacéuticas informaron que habían gastado una estimación de 25,3 millones de euros anuales en cabildeo mientras desplegaban 290 grupos de presión para influir en las políticas de la UE. Un portavoz de Janssen, la filial de investigación y desarrollo farmacéutico que desarrolló la vacuna de una sola inyección de Johnson & Johnson, fue aún más allá al supuestamente amenazar a la oficina del Primer Ministro belga de que la compañía reconsideraría sus inversiones en el país si Bélgica respaldaba la exención.
(Véase también: Un informe de STOP AIDS basado en investigaciones en Europa: Acceso denegado – Lo que sucede cuando Big Pharma está en el asiento del conductor).
Si bien Estados Unidos cambiaría su postura y eventualmente apoyaría una exención limitada solo aplicable a las vacunas, adoptada a través de la Decisión Ministerial de la OMC el 17 de junio de 2022, los esfuerzos de Big Pharma ya habían dado sus frutos, ya que la implementación de cualquier exención se retrasó significativamente y su alcance se redujo sustancialmente.
Después del cambio de política de los Estados Unidos, que ocurrió después de la transición de la administración Trump a la administración Biden, los esfuerzos de cabildeo, impulsados en gran medida por entidades opuestas a la exención, continuaron. En 2022, el año en el que se contrataron más grupos de presión, las entidades que se opusieron a la exención superaron en número a las contratadas por los partidarios 32 a 1.
Además, las revelaciones de cabildeo muestran un esfuerzo de oposición que se extiende hasta 2024. Dos docenas de entidades revelaron el cabildeo sobre la exención hasta la primera mitad de 2024, cuando concluyeron las conversaciones sobre la exención de los ADPIC. La mayoría de estas entidades eran empresas farmacéuticas o de biotecnología y asociaciones comerciales que representaban a las industrias. Pero los esfuerzos de la industria fueron más allá de la exención en sí.
influencia de la industria en las negociaciones de acuerdos de pandemia en curso
Insatisfecha con la mera eliminación de la exención en la OMC, la industria ha seguido adelante con sus esfuerzos para socavar aún más en las negociaciones en curso la equidad global en el Acuerdo de Pandemia de la OMS.
Sus esfuerzos de cabildeo parecen haber tenido un impacto duradero, ya que muchas naciones ricas involucradas en las negociaciones del Acuerdo sobre la Pandemia de la OMS han adoptado desde entonces posturas que son paralelas a las que la industria mantuvo anteriormente en la OMC. Países como los Estados Unidos y los estados miembros de la UE, por ejemplo, han adoptado posiciones estrictas en torno al intercambio de tecnología y sobre la flexibilidad de la propiedad intelectual, haciéndose eco de los puntos de discusión de la industria, que afirman que ciertas disposiciones para hacer operativa la equidad implicarían «eliminar las protecciones de la propiedad intelectual».
Además de la exención de TRIPS, las asociaciones comerciales que representan a la industria farmacéutica y biotecnológica también han presionado al gobierno de los Estados Unidos sobre el Acuerdo de Pandemia. Estas incluyen algunos de los grupos comerciales más poderosos del país, la Cámara de Comercio de los Estados Unidos y la Organización de Innovación Biotecnológica. En 2024, la Cámara de Comercio envió 49 grupos de presión al Capitolio para presionar sobre el Acuerdo de Pandemia. La Organización de Innovación Biotecnológica desplegó 16 grupos de presión para influir en las negociaciones del Acuerdo Pandémico, según la divulgación más reciente de su cabildeo.
Aunque muchos países ricos tienen a su disposición amplias herramientas jurídicas y reglamentarias para obligar a la industria a compartir tecnología y conocimientos técnicos por medios no voluntarios, no se puede exagerar la hipocresía de luchar por un lenguaje que pueda disuadir a los países de ingresos bajos y medios de hacer lo mismo. Además, algunos de estos países ricos también se oponen a la inclusión de una «cláusula de paz» que reafirmaría un compromiso colectivo de los signatarios de abstenerse de «cualquier presión directa o indirecta» para desalentar el uso del Acuerdo sobre los ADPIC y otras flexibilidades previstas en el Artículo 9, una disposición relacionada con la investigación y el desarrollo. pesar de haber sido reiniciadas en Ginebra el pasado septiembre, las negociaciones no han superado los desacuerdos críticos que existen a lo largo de la división Norte-Sur sobre cómo abordar áreas críticas para la equidad, incluido el Sistema de Intercambio de Beneficios y Acceso a Patógenos propuesto (PABS) y las disposiciones sobre la transferencia de tecnología y conocimientos para la producción de productos de salud relacionados con la pandemia. Los países de altos ingresos continúan oponiéndose a avances significativos en el PABS y presionando por incluir disposiciones vinculantes que definirían la transferencia de tecnología en términos de «condiciones voluntarias» y «acordadas mutuamente», la posición preferida de la industria que está siendo defendida fuertemente por los países en su mayoría desarrollados y otros actores con sede en Ginebra.
Aunque muchos países ricos tienen a su disposición amplias herramientas jurídicas y reglamentarias para obligar a la industria a compartir tecnología y conocimientos técnicos por medios no voluntarios, no es exagerado destacar la hipocresía de plantear un lenguaje que pueda disuadir a los países de ingresos bajos y medios de hacer lo mismo. Además, algunos de estos países ricos también se oponen a la inclusión de una «cláusula de paz» que reafirmaría un compromiso colectivo de los signatarios de abstenerse de «cualquier presión directa o indirecta» para desalentar el uso del Acuerdo sobre los ADPIC y otras flexibilidades previstas en el Artículo 9, una disposición relacionada con la investigación y el desarrollo.
Teniendo en cuenta las complejidades de estas negociaciones globales entre países con capacidades y circunstancias muy diferentes, es fácil perder de vista la influencia dañina que el lobby farmacéutico ha tenido en el proceso. Sin embargo, la cuestión fundamental que se interpone en el camino del gran acuerdo que puede prevenir brotes mortales y poner a disposición herramientas médicas para combatirlos, es si las naciones ricas están preparadas para romper el acuerdo con la industria farmacéutica y decir «no» a la codicia insaciable y los intereses corporativos.
Los fabricantes de medicamentos y otros actores tienen un interés financiero en mantener la propiedad de las herramientas médicas y el conocimiento, solo están dispuestas a compartir utilizando estrictamente sus propias reglas, incluso durante las catástrofes globales, y están haciendo todo lo posible para proteger su capacidad de ganancias sin ser agobiados por las preocupaciones sobre la equidad.
El diario El País publicaba a finales de septiembre una carta abierta de cuatro reconocidos investigadores internacionales que han estado desarrollando investigaciones sobre la enfermedad de Alzheimer, donde solicitaban que la EMA reconsiderara su decisión sobre la aprobación de lecanemab. Efectivamente los autores señalaban que la EMA “niega el acceso a los pacientes y sus médicos a un tratamiento que puede cambiarles la vida”.
El debate sobre el anticuerpo monoclonal Lecanemab para Alzheimer en fase temprana, es importante y ha sido motivo de fuerte controversia. Public Citizen se ha hecho eco de una información de un medio generalista importante como el The New York Times que mostraba el rechazo a la aprobación dedeterminados grupos de profesionales médicos que han puesto en duda los ensayos clínicos.Los estudios publicados constatan su efectividad clínica (27% de pacientes en los que se observa un retraso en la evolución de la enfermedad) y su alto coste (25.000 € paciente/ año). Sin embargo, su alto coste y muy moderado beneficio han motivado que algunas administraciones públicas, como en el Reino Unido, decidieran la no financiación de esta medicación.
Es una discusión de notable calado, donde se incluyen también reflexiones desde el punto de vista de la ética clínica. Pero además introduce aún más dudas en el proceso de decisión de financiación del fármaco.
Nuño Domínguez señalaba en El País del 25 de septiembre pasado: “El primer fármaco que ha mostrado algún efecto contra el alzhéimer en varias décadas —el lecanemab— está dividiendo a la comunidad científica y médica. Estados Unidos, China, Japón, Israel, Corea del Sur o Emiratos Árabes ya han aprobado el uso de este anticuerpo monoclonal. Reino Unido le acaba de dar el visto bueno, pero su sistema público no lo suministrará, porque considera que sus efectos son demasiado modestos para su coste, de unos 24.000 euros al año por paciente. A contracorriente, la Agencia Europea del Medicamento (EMA)lo rechazó en julioal considerar que sus beneficios —reduce un 27% el avance de la enfermedad— no compensan los riesgos —hemorragias cerebrales y muerte de dos pacientes—. (…) En un movimiento poco habitual, cuatro prestigiosos investigadores internacionales que llevan décadas estudiando esta enfermedad han enviado una carta abierta a EL PAÍS para pedir que Europa reconsidere su decisión y apruebe esta clase de fármacos. La decisión de la EMA “niega el acceso a los pacientes y sus médicos a un tratamiento que puede cambiarles la vida”, escriben Bart de Strooper, cofundador del Instituto de Investigación de Demencia de Reino Unido, Henrik Zetterberg, de la Universidad de Gotemburgo (Suecia), Christian Haass, de Múnich (Alemania), y John Hardy, del University College (Reino Unido)”.
En sentido contrario, desde Public Citizen, el 23 de octubre sembraban dudas sobre los ensayos clínicos que han conducido a la aprobación: ”Un inquietante artículo del New York Times el miércoles reveló que, durante los ensayos clínicos de dos medicamentos recientemente aprobados para la enfermedad de Alzheimer, a los participantes no se les dijo si sus perfiles genéticos aumentaban sus riesgos de lesiones cerebrales al recibir los medicamentos. Según el artículo, en los ensayos de lecanemab (Leqembi) y donanemab-azbt (Kisunla) «los voluntarios primero tuvieron que firmar formularios de consentimiento que decían que las personas con ciertos perfiles genéticos enfrentaban mayores riesgos de lesiones cerebrales al recibir el medicamento, y que los participantes se harían a pruebas para ellos ¾, pero no se les diría los resultados».
Doctor Robert Steinbrook, director del Grupo de Investigación de Salud del Ciudadano Público, pide a la Administración de Alimentos y Medicamentos (FDA) y a la Oficina de Protección de la Investigación Humana (OHRP) del Departamento de Salud y Servicios Humanos (DHHS) que investiguen la realización de los dos ensayos y las acciones de las juntas de revisión institucional que aprobaron las disposiciones de secreto. Emitió la siguiente declaración:
«El requisito bajo las regulaciones federales de obtener el consentimiento informado antes de involucrar a las personas en la investigación es una protección fundamental para los sujetos de investigación. Las revelaciones sobre las disposiciones de secreto en los ensayos de la enfermedad de Alzheimer sugieren que el principio del consentimiento informado se socavó. Si los sujetos hubieran sabido que tenían un mayor riesgo de lesiones cerebrales, podrían haber decidido no participar en los ensayos. Esta es la información que los posibles participantes querrían saber».
La FDA y la OHRP deberían investigar formalmente la realización de los dos ensayos y las acciones de las juntas de revisión institucional con supervisión de los estudios. Tanto el lecanemab como el donanemab-azbt son medicamentos con beneficios clínicos muy modestos que no superan sus riesgos sustanciales de seguridad, incluida la inflamación y el sangrado cerebral. La FDA no debería haber aprobado ninguno de los medicamentos».
Comparte en tus Redes
Uso de cookies
Este sitio web utiliza cookies para que usted tenga la mejor experiencia de usuario. Más info