ORIGINAL. Revista nº 42 Octubre 2025
Luz de Myotanh Vázquez Canales.
Médica de Familia y Comunitaria en el Consultorio Estivella (Estivella, Valencia). Coordinadora del Grup del Medicament de la SoVaMFiC. https://grupmedicament.wordpress.com/
Ponencia presentada en el Seminario de Innovación en Atención Primaria 2025, “Medicamentos, salud y sistemas sanitarios”, celebrado en Madrid los días 23 y 24 de septiembre.
Relato y contexto
El colectivo sanitario, y en especial la Medicina, se ve envuelto en un contexto donde la farmaindustria (FI) está presenta de forma directa o indirecta. A veces, es tan inevitable que en tu almuerzo de 15 minutos hay un café pagado que no se ha elegido. La FI forma parte de la coyuntura y la idiosincrasia de los centros sanitarios públicos. Los adjuntos y los residentes en formación se ven envueltos sin cuestionarse, en muchas ocasiones, la ética de que esto suceda. Ejemplificaré una situación basada en hechos reales de lo que puede suceder una mañana cualquiera en un centro de salud.
María acaba de llegar a su centro de salud. Es su primer día de R1. A la entrada, junto al mostrador, hay unos señores con chaquetas y maletines que desentonan ligeramente con la estética habitual del centro. Ve un pequeño círculo de médicas rodeando a estas personas. Todos parecen sonrientes y muy educados. De sus maletines sacan panfletos, bolígrafos y alguna cosa más que entregan a las médicas del centro. María decide pasar de largo y sube a buscar a su tutora. No está. Vuelve a bajar. Entre el círculo de médicas, la encuentra. Su tutora amablemente decide introducirla a todos los señores del maletín. Uno de ellos decide invitarlas a desayunar. Son las 8,15h y la consulta no empieza hasta las 9h. Nadie parece cuestionar nada y todo el mundo ve normal ir al desayuno.
Acuden a un barecito cerca del centro de salud donde la camarera ya conoce a Joaquín, el representante. Joaquín entabla una conversación agradable y pregunta a María por sus gustos en música, restaurantes y si ha visto la última serie de Netflix, Wednesday, donde Jenna Ortega sigue igual de magnífica que en la primera temporada. Su tutora y ella piden unas tostadas con un café con leche y Joaquín, un café solo. El representante le ofrece a María un acto de bienvenida de residentes en el que primero hablará el jefe de servicio de Neumología de su hospital de referencia y luego harán un ágape. Su tutora la anima a asistir, pues la charla tiene buena pinta y luego podrá disfrutar de un cóctel en uno de los mejores rooftop de la ciudad con sus compañeros de promoción. María se siente entusiasmada con la idea y da las gracias a su tutora y a Joaquín por la invitación. Al acabar el desayuno María hace un ademán de pagar su parte, pero Joaquín se les ha adelantado sin que ella se diera cuenta. Llegan 5 minutos tarde a la consulta. Todos los pacientes de las 9 a las 9.15h esperan puntuales con una sonrisa y dan los buenos días. María no deja de pensar en el evento de Joaquín y todo lo que va a aprender sobre inhaladores para el EPOC.
En el ejemplo expuesto se pueden identificar una serie de elementos a los que he llamado factores de poder que influyen en la permanencia de la FI (Figura 1).
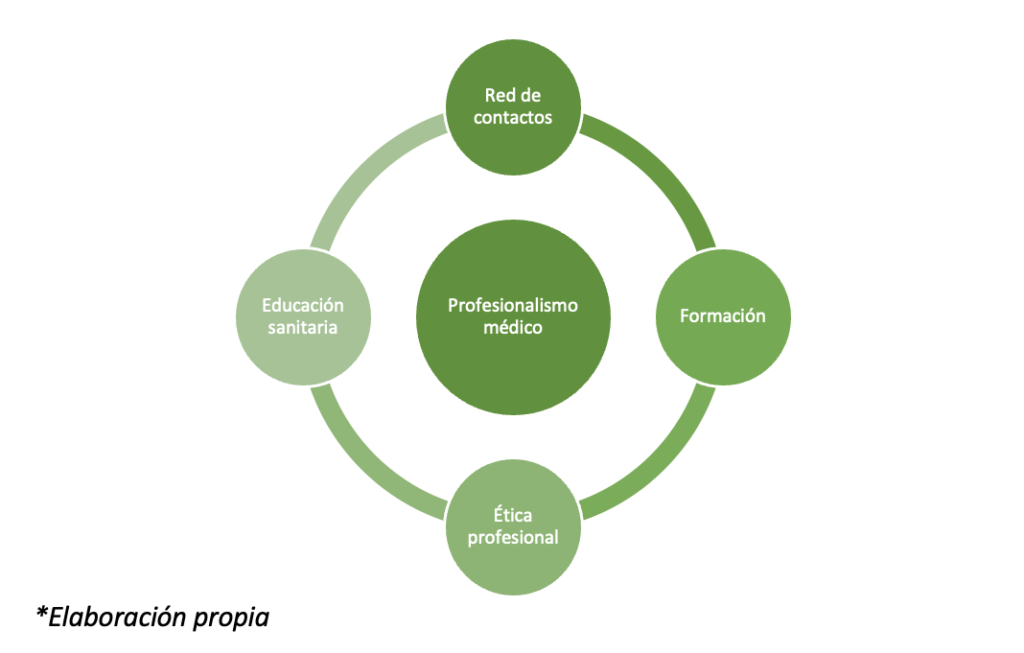
- La red
La década los 90 supuso un cambio en el paradigma de rentabilizar las industrias farmacéuticas. La inversión en I+D pasó a un segundo plano, y el marketing y la inversión en publicidad encumbraron una parte importante del presupuesto de la FI pues ofrecía más beneficios. Esto ha tenido graves consecuencias en los sistemas de salud, dado que las instituciones han pasado a ser espacios donde se negocia la compra/venta de fármacos y tecnologías1.
Víctimas y verdugos conviven en un mismo espacio, pues no deja de ser una forma de hacer corrupción aceptada e integrada en instituciones públicas donde la transparencia está prácticamente ausente. La RAE establece en una de sus acepciones sobre el término corrupción “en las organizaciones, especialmente en las públicas, prácticas consistentes en la utilización indebida o ilícita de las funciones de aquellas en provecho de sus gestores”; es llamativo cómo vincula el término corrupción con las instituciones públicas y los gestores que conviven en ellas. Un ejemplo bien documentado de esta situación es la publicación que realizó Ángel Martín “La red oscura que las multinacionales farmacéuticas ocultan tras pagos a profesionales sanitarios: sus lideres de opinión en el SNS y su red de intereses al descubierto2”. En él hace una revisión minuciosa de la red social de las multinacionales farmacéuticas y sus pagos a sanitarios, a los que considera líderes claves de opinión (KOL, en inglés). Estos KOL suelen ser médicos que ocupan importantes cargos de gestión dentro del Sistema Nacional de Salud (SNS) como jefaturas de servicio, sección e inclusive gestores o directores de hospital. Los pagos que perciben suelen oscilar entre 5.000 a 150.000 euros procedentes de varias multinacionales. Además, este salario extra anual no lo declaran de forma directa en los portales de transparencia de las instituciones en las que trabajan. Las especialidades médicas cuyos KOL reciben más pagos son oncología, hematología, reumatología y dermatología. La Atención Primaria (AP) no está exenta de esta red, pero las sumas de dinero que reciben sus líderes de opinión son menos cuantiosas que las especialidades mencionadas. Los KOL de AP suelen obtener beneficios procedentes de las multinacionales dedicadas a las vacunas y los antidiabéticos orales.
- El aprendizaje supersticioso (Superstitious learning)
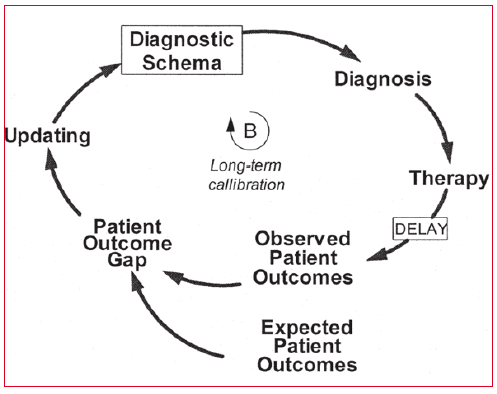
El modelo de Berner y Graber3 explica que en condiciones óptimas el proceso de aprendizaje del médico pasa por distintas fases. La primera condición es que el médico tiene una serie de esquemas mentales que desde el punto de vista cognitivo viene a ser todos aquellos constructos algorítmicos que se encuentran interrelacionados entre sí y pertenecen a la materia bajo la que se han especializado. Estos esquemas son sumamente importantes porque son los que ayudan a la toma de decisiones cuando se integran junto la información que el paciente transmite. Este proceso de integración permitirá realizar un diagnóstico y, en base a esto, proponer un tratamiento. Cuando se aplica un determinado tratamiento suceden dos situaciones que se dan casi al mismo tiempo: uno, son los resultados esperados y otro, los resultados observados. Y en este impass que denominan gap, es donde como sanitarios hacemos una actualización/aprendizaje mediante la propia observación y/o búsqueda de información sobre ese caso. Esto actualiza los esquemas mentales que posteriormente ayudarán a hacer mejores diagnósticos y a atender mejor al paciente en las sucesivas visitas (Figura 2).
Figura 2. Cuando la calibración funciona: un proceso de retroalimentación que funciona
Rudolph JW, Morrison JB. Sidestepping Superstitious Learning, Ambiguity, and Other Roadblocks: A Feedback Model of Diagnostic Problem Solving. American Journal of Medicine [Internet]. 2008 May 1 [cited 2025 Aug 23];121(5 SUPPL.):S34–7. Disponible en: https://www.amjmed.com/action/showFullText?pii=S000293430800154X
Pero, la retroalimentación observada en la Figura 1 se puede ver alterada por tres situaciones, según Rudolph y Morrison3: retroalimentación ambigua, retrasos en los resultados del paciente y el aprendizaje supersticioso. Me centraré en este último, por la relevancia que tiene en cómo la FI es capaz de modificar este proceso.
El aprendizaje supersticioso son explicaciones basadas en la confianza y conocimientos propios que no se encuentran sujetas a la evidencia clínica. El aprendizaje supersticioso está sustentado en el sesgo de autoconfirmación que se define como:
Es un atajo mental involuntario que nos lleva a buscar, interpretar y recordar información que apoya nuestras creencias y opiniones preexistentes, mientras ignoramos o restamos importancia a la información que las contradice.
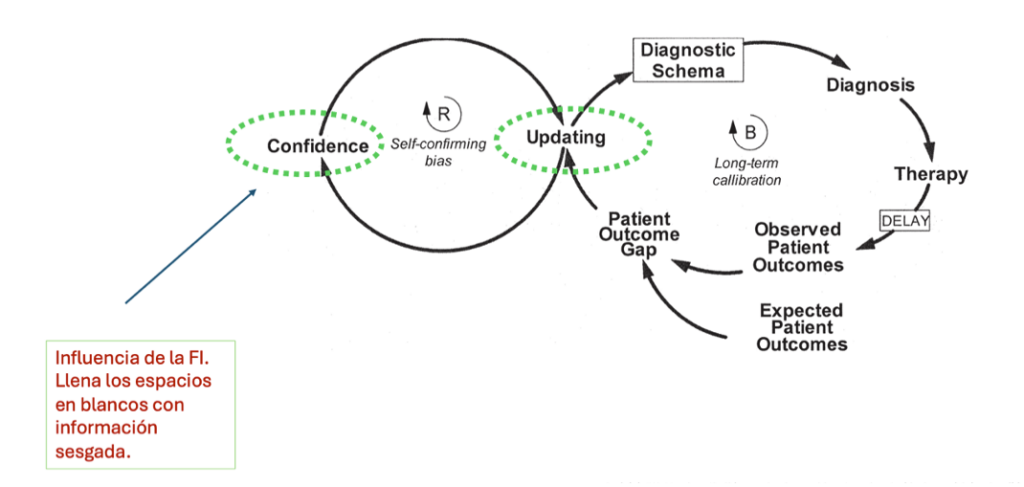
Este sesgo cognitivo puede afectar a la toma de decisiones en diversos ámbitos, desde la vida cotidiana y las redes sociales hasta la investigación científica y médica, llevando a conclusiones erróneas o menos informadas. Esto puede llegar a ser peligroso en una profesión como es la médica, dado que la toma decisiones no puede basarse exclusivamente en creencias propias o no contrastadas. Un ejemplo de ello es cuando los representantes ofrecen información sobre un determinado medicamento, el objetivo es la venta de este, por lo que mucha de la información que ofrecen está sesgada. Van a enseñar los resultados de aquellos estudios estrictamente seleccionados por sus buenos resultados, y nunca enseñarán las investigaciones menos favorables. Si como sanitarios no contrastamos la información que ofrecen con fuentes fiables de evidencia, la actuación puede ser errónea o menos precisa. Berner y Graber3 demuestran que el sesgo de autoconfirmación se cumple sistemáticamente y que los sanitarios no suelen comparar la información que reciben con otras fuentes. Además, afirman que en la medida en la que aumenta la confianza propia, la necesidad de actualizarse disminuye con la aparición del sesgo de autoconfirmación. Y precisamente, la mejora en el aprendizaje y la utilización de evidencia científica permite a los clínicos responder de forma más adecuada a la resolución de problemas con el tiempo (Figura 3). Algunos estudios hablan de que solamente un 15% de las decisiones médicas se basan en la evidencia.
Figura 3. Como la confianza impide calibrar de forma adecuada la toma de decisiones diagnósticas
Rudolph JW, Morrison JB. Sidestepping Superstitious Learning, Ambiguity, and Other Roadblocks: A Feedback Model of Diagnostic Problem Solving. American Journal of Medicine [Internet]. 2008 May 1 [cited 2025 Aug 23];121(5 SUPPL.):S34–7. Disponible en: https://www.amjmed.com/action/showFullText?pii=S000293430800154X
- Congresos y conferencias, ¿sirven para algo?
Los congresos, conferencias, workshops o simposios son prácticamente imposible de cuantificar al año. J. Ionnadis4 estima que puede ser que haya más de 100.000 al año. Además, se cuestiona si este tipo de encuentros ofrecen conocimiento, más allá de la huella ecológica que genera que cientos y miles de personas se desplacen a una ciudad.
La evidencia acumulada a lo largo de los años sugiere que las reuniones médicas pueden servir a un sistema de valores perjudiciales para la medicina y la atención sanitaria. Esta afirmación se sustenta en algunas cuestiones que suceden en los congresos:
- Los trabajos científicos que se presentan suelen ser resúmenes de escasas palabras que no llegarán la mayoría de ellos a artículos científicos. Sin embargo, sí que contribuirá a elaborar un currículum vitae inflado de publicaciones de escaso valor.
- Una parte importante de los conferenciantes son líderes de opinión cuyas “valiosas” declaraciones pueden ejercer una amplia influencia, independientemente de la evidencia, en ausencia de ella o incluso contra ella.
- Las grandes sociedades científicas de profesionales, en muchas ocasiones, no eligen a sus KOL por el liderazgo, el mérito científico, el trabajo duro y la originalidad del pensamiento, sino más bien por la capacidad de navegar en los círculos de poder.
- Los programas científicos suelen figurar ponentes con conflictos de interés, que no suelen declarar. Así, la información ofrecida tiene una alta probabilidad de estar sesgada.
Quizás, ante esta incertidumbre y discrepancia sobre la utilidad o no de los congresos cabría plantearse la realización de estudios formales para evaluar la pertinencia de seguir con este tipo de eventos y los métodos de difusión de la investigación y educación con el objetivo de mejorar la formación.
- La ética profesional: el profesionalismo
“Yo recibo a la FI pero sé perfectamente que esto no va a influirme en nada”; “Sé que no los debería de recibir a los visitadores, pero me sabe mal, a fin de cuentas, están haciendo su trabajo y no tienen la culpa nada”. (Frase de una jefa de servicio de un hospital público)
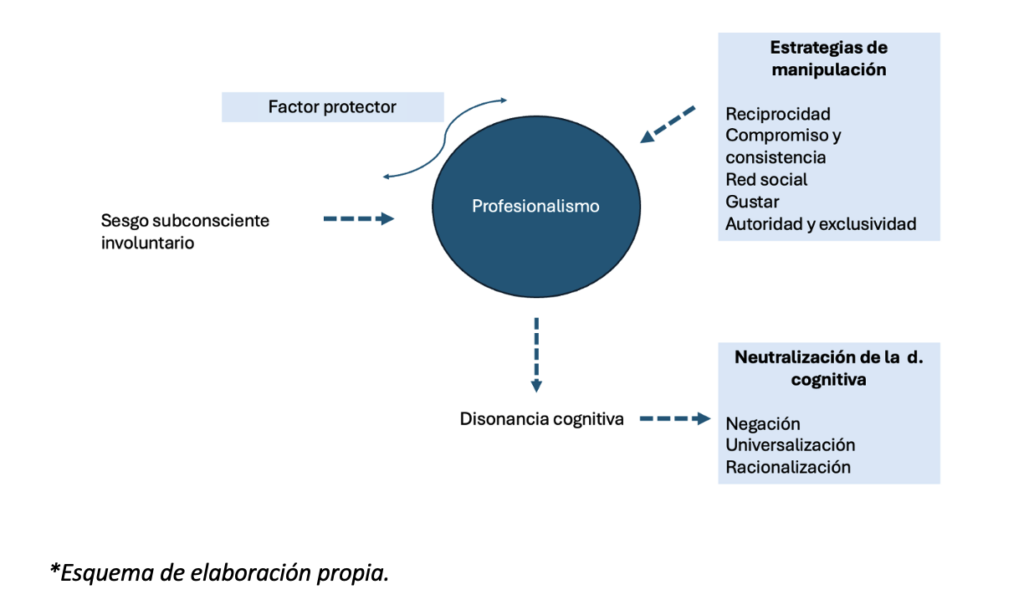
Los incentivos que reciben los profesionales procedentes de la FI suelen ser económicos (pago de congresos, viajes, comidas) y no económicos (trato preferente). La mayor parte de profesionales sanitarios no reconocen la vulnerabilidad que tienen frente a las estrategias comerciales y los pagos. La mayoría creen que son inmunes y que no les afecta en la toma de decisiones, pero curiosamente sí creen que la FI influye en sus compañeros. Las estrategias de marketing que utilizan los visitadores hacen que la información sea más verosímil por lo que los sanitarios son más vulnerables1. Las 8 estrategias más utilizadas son: simpatía, autoridad, aceptación social, unidad, escasez, reciprocidad, coherencia/compromiso y presión emocional. Esta última se utiliza casi 3 veces más que las demás5. En este sentido, el profesionalismo médico puede ayudar a evitar la corrupción intencionada pero no puede evitar aquello que se conoce como el sesgo subconsciente involuntario, que hará que se dejen seducir.
La aceptación de los incentivos, sumado a sus tácticas de marketing y la frecuencia con la que los visitadores acuden a vender sus productos, genera una disonancia cognitiva que viene a ser el malestar que surge de la discrepancia entre creencias y comportamientos. La disonancia cognitiva se puede neutralizar con estrategias como1:
- Negación. Ej. No pasa nada porque reciba el pago por la inscripción a un congreso.
- Universalización. Ej. Todo el mundo acude a congresos y conferencias pagadas por la industria, pues yo también lo puedo hacer.
- Racionalización. Ej. Con lo mal que nos trata la administración pública, merezco que la FI me pague un extra para irme de vacaciones con la familia.
Por tanto, a pesar de la potente influencia protectora que otorga el profesionalismo, se ve mermado. Así, es necesario que los sanitarios se familiaricen con estas estrategias para poder evitar esta manipulación consciente de la FI.
*Esquema de elaboración propia.
Conclusiones
- La red con que actúa la FI se encuentra perfectamente enmarañada dentro de los sistemas sanitarios. El mal es endémico y la FI se ha integrado en la coyuntura sanitaria sin que nadie cuestione sus actuaciones.
- Las estrategias de marketing utilizadas son capaces de manipular la forma en la que se aprende hasta el punto de dar por sentado creencias que no están basadas en la evidencia.
- Los congresos son espacios de poder donde solamente unos pocos, los KOL, pueden brillar, aupados por una industria que no valora el buen hacer.
- La falta de identificación de las estrategias de manipulación por parte de los profesionales supone una merma del profesionalismo y la ética profesional.
Bibliografía
- Novoa A, Gérvas J, Ponte C. Salvaguardas, deriva institucional e industrias farmacéuticas. AMF Actualización en Medicina de Familia [Internet]. 2012 [citado 2025 Aug 19]; Disponible en: https://amf-semfyc.com/es/web/articulo/salvaguardas-deriva-institucional-e-industrias-farmaceuticas
- Martín ÁM. La red oscura que las multinacionales farmacéuticas ocultan tras los pagos a profesionales sanitarios: sus líderes de opinión en el SNS y su red de intereses al descubierto – Asociación Acceso Justo al Medicamento [Internet]. Asociación Acceso Justo al Medicamento. 2024 [citado 2025 Aug 19]. Disponible en: https://accesojustomedicamento.org/la-red-oscura-que-las-multinacionales-farmaceuticas-ocultan-tras-los-pagos-a-profesionales-sanitarios-sus-lideres-de-opinion-en-el-sns-y-su-red-de-intereses-al-descubierto/
- Rudolph JW, Morrison JB. Sidestepping Superstitious Learning, Ambiguity, and Other Roadblocks: A Feedback Model of Diagnostic Problem Solving. American Journal of Medicine [Internet]. 2008 May 1 [cited 2025 Aug 23];121(5 SUPPL.):S34–7. Disponible en: https://www.amjmed.com/action/showFullText?pii=S000293430800154X
- Ioannidis JPA. Are Medical Conferences Useful? And for Whom? JAMA [Internet]. 2012 Mar 28 [citado 2025 Aug 23];307(12):1257–8. Disponible en: https://jamanetwork.com/journals/jama/fullarticle/1105123
- Dankers M, Verlegh P, Weber K, Nelissen-Vrancken M, van Dijk L, Mantel-Teeuwisse A. Marketing of medicines in primary care: An analysis of direct marketing mailings and advertisements. PLoS One [Internet]. 2023 Aug 1 [citado 2025 Aug 23];18(8):e0290603. Disponible en: https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0290603
Preguntas
- ¿Cómo se puede mejorar la influencia de la farmaindustria en el contexto sanitario?
- ¿Por qué un número importante de sanitarios cree ser inmune a la farmaindustria y no se cuestiona su influencia?
- ¿Cómo se debería de gestionar por parte de las conserjerías de sanidad los incentivos que los sanitarios reciben teniendo en cuenta que están colaborando con multinacionales privadas y recibiendo sobresueldos que, a veces, sobrepasan el propio salario bruto/anual?