Andreu Segura es epidemiólogo, médico especialista en Medicina Preventiva y Salud Pública. Con una dilatada experiencia en el mundo de las asociaciones profesionales. Fue fundador de la Sociedad Española de Epidemiología (SEE) y de la Sociedad Española de Salud Pública y Administración Sanitaria (SESPAS). Ha sido profesor en la Universidad de Barcelona. Ha trabajado en Salud Pública fundamentalmente en Cataluña.
El número 45 de la revista ACCESO JUSTO AL MEDICAMENTO, https://accesojustomedicamento.org/revista-de-aajm/ ya está disponible. El denominador común del presente número de la revista AJM es la imperativa necesidad de anteponer el interés público y el derecho universal a la salud frente al modelo de monopolios de la industria farmacéutica. Esta idea central se manifiesta de diversas formas en los textos. El medicamento como bien social, no solo comercial. El editorial de Jaume Vidal, senior Policy Advisor, Health Action International, advierte sobre los riesgos interconectados por las decisiones de los EE.UU. y la deriva mercantilista de la Unión Europea, aspecto, este último que analiza Fernando Lamata, presidente de la Comisión Editorial de esta revista, cuando nos presenta las penosas condiciones aprobadas en el acuerdo de la OMS sobre pandemias. Raúl Calvo Rico, presidente del Colegio Oficial de Médicos de Toledo, denuncia cómo la industria influye en todas las etapas: desde la formación ética de los estudiantes de medicina y las decisiones de prescripción, hasta la presión sobre los organismos reguladores (como la EMA) y los líderes políticos. Felipe de la Fuente, farmacéutico, argumenta que la política debe dejar de ser una facilitadora de la rapidez comercial para convertirse en un “motor de acceso justo» que defina y exija verdadera innovación basada en el beneficio social. Entre los materiales originales, finalmente, Antonio Pujol de Castro, MIR de Medicina Preventiva y Salud Pública, pone el acento en los determinantes de la salud y explica la influencia del precio de los medicamentos: “en España , un 4,6% de la población (unos 2,2 millones de personas) declaró en 2025 no haber podido tomar la medicación recetada por motivos económicos. El copago -apostilla- está expulsando a los más vulnerables. En definitiva, los autores abogan por un cambio de paradigma legislativo y ético que recupere la soberanía de la salud pública, garantizando la transparencia en los costes, la independencia en la formación médica y un modelo de I+D orientado a las necesidades reales de la población y no solo a la rentabilidad corporativa. En la recopilación de artículos por otras fuentes, en la rAAJM 45, se recogen dos publicaciones deseadas de Public Citizen. La primera se refiere a que más de 100 organizaciones instan a los países a resistir los ataques comerciales de Trump y proteger el acceso a medicamentos asequibles, la segunda, denuncia que el acuerdo de Trump con Argentina marca un mínimo en el comercio favorable a las grandes farmacéuticas. La revista se hace eco, asimismo, de una información de Peopless Health Dispatch, relativa aquelarre la coalición sanitaria de Indonesia exige el regreso de las leyes que les protegían frente a reverdecimiento de las patentes por parte de las grandes corporaciones. Finalmente, reproducimos una reflexión de singular claridad del analista Dean Baker, sobre la falsedad de la política de precios de los medicamentos del presidente de los EE.UU, que desde su punto de vista, está haciendo a las Big Pharma más ricas. Para los que utilicen la versión en PDF, accesible igualmente desde el enlace a la página web de la AAJM señalado al principio, recomendamos la lectura de los 4 informes y documentos seleccionados por la Comisión de Redacción. Como cierre de la revista 45 de la AAJM, en nuestra sección “Maldigo la poesía”, traemos la “Oda a los niños de Madrid muertos por la metralla”, escrita por el premio Nobel español Vicente Aleixandre en 1937, y vemos su desgraciado y terrible paralelismo con los niños asesinados en los bombardeos sobre la franja de Gaza (imagen que muy lamentablemente se puede extender en estos días a todo Oriente Próximo). Vídeo resumen de la revista elaborado por la IA de NotebookLM: PINCHANDO AQUÍ Acceso al PDF del Nº 45 de la revista AJM, en:
Consultor independiente. Miembro de la Comisión Editorial de la rAJM.
Vivimos tiempos extraordinarios, de final de etapa y principios inciertos de algo diferente, distinto. Un periodo donde las oportunidades conviven con los riesgos. Desde los desafíos que enfrenta la institucionalidad y práctica de la salud global a las ambigüedades en la transformación del ámbito regulatorio farmacéutico europeo o los retos por completar una ambiciosa agenda farmacéutica y sanitaria a nivel doméstico. Procesos paralelos, pero con múltiples puntos de contacto; desarrollos y desenlaces que en cada nivel pueden afectar en mayor o menor medida a otros escenarios.
Recientes decisiones del gobierno estadounidense como la salida intempestiva de la Organización Mundial de la Salud (OMS) o la súbita suspensión de programas de asistencia vital como el President’s Emergency Plan for AIDS Relief (PEPFAR) han erosionado los pilares institucionales y financieros de la salud global. La incapacidad (por motivos de muy distinta naturaleza) del resto de actores a responder, compensar o paliar el impacto de estas acciones unilaterales es si cabe aún más preocupante; nos obliga a interrogarnos sobre la viabilidad de un modelo nacido tras la 2ª Guerra Mundial y consolidado tras el final del enfrentamiento entre bloques.
La Unión Europea (UE) no va a erigirse en salvadora de un orden internacional basado en la relación entre iguales, guiado por la solidaridad y la colaboración ante la amenaza compartida. Los discursos y gestos desde Bruselas (y aplaudidos por la mayoría de las capitales) apuntan al repliegue, rearme y reindustrialización con la acción en salud supeditada a la agenda comercial y mercantil. Las concesiones a la industria en dosieres claves como la reforma de la legislación farmacéutica o la escasez de medicamentos y las posiciones duras (cuasi recalcitrantes) en la negociación del acuerdo de pandemias por parte de la Comisión Europea, en nombre de los estados miembros, son algunos ejemplos de la deriva comunitaria.
Es en los panoramas de desolación donde la resistencia cobra valor. Son pocos los gobiernos que, en los últimos años, han hecho una apuesta firme por la Salud tanto en el ámbito interno como allende las fronteras. Con aciertos y errores, el ejecutivo español lo ha intentado, no con pocos éxitos: el retorno al Comité Ejecutivo de la OMS, la aprobación en la Asamblea Mundial de la Salud de la resolución con el primer plan estratégico contra enfermedades raras o la profundización en la cooperación regulatoria de productos farmacéuticos con las Americas son hitos indiscutibles; otras contribuciones, más calladas y menos visibles, también han sido valoradas en foros y espacios. A nivel interno, la realidad es tozuda y el proyecto de nueva Ley del Medicamento, una asignatura estructuralmente pendiente, se encuentra en el limbo legislativo de mayorías fluctuantes; en este caso la voluntad política no es suficiente para alcanzar los objetivos.
En resumen, tiempos de cambio e incertidumbre, pero también de esperanza necesaria y optimismo militante; es el momento de continuar esforzándonos y colaborando para que la salud no dependa de ingreso ni frontera y el acceso a medicamentos sea derecho y no quimera.
Según la Nota informativa de Third World Network, en relación con la 5ª reunión del Grupo de Trabajo Intergubernamental (IGWG5), llevada a cabo en la sede de la OMS en Ginebra entre el 9 y el 14 de febrero, los países del Norte, con la Unión Europea de forma destacada, se oponen a un sistema de Acceso a Patógenos y Distribución de Beneficios (PABS) equitativo y justo (“OMS: la UE retrocede en los compromisos del acuerdo sobre la pandemia e ignora evidencias y precedentes. Third World Network, 20 febrero 2026).
Recordemos que, en mayo de 2025, la OMS alcanzó un acuerdo para hacer frente a futuras pandemias (Emergencias de Salud Pública de Importancia Internacional). Como ya analizamos en rAJM, ese acuerdo era muy insuficiente y, además, su entrada en vigor se demoraba un año hasta que se aprobara el anexo sobre el sistema PABS.
Pues bien, durante estos meses, los países ricos, incluyendo la UE, están tratando de imponer un sistema PABS injusto. Así, la cesión (por los países en desarrollo) de patógenos con potencial pandémico, y de los datos de secuencias genéticas, sería obligatoria; pero la firma de contratos de las empresas (y países desarrollados) receptores de la información para garantizar una distribución equitativa de las posibles vacunas, tratamientos o medios diagnósticos, sería voluntaria. Es decir, que no se les podría obligar a ceder licencias, ni a transferir tecnología, ni a facilitar reservas estratégicas en todas las regiones. Es una propuesta desequilibrada que no facilita la gobernanza global para hacer frente a futuras pandemias (que vendrán antes que después) y consolida las desigualdades estructurales actuales.
Conviene insistir en la necesidad de un cambio de modelo para la investigación, desarrollo y distribución de vacunas, tratamientos y medios diagnósticos. El modelo actual basado en patentes y derechos de propiedad intelectual, que conceden monopolios de producción y distribución a las empresas, genera enormes beneficios para unos pocos y deja sin acceso a medicamentos a millones de personas. Lo mismo ocurrirá en futuras pandemias.
La I+D de medicamentos y vacunas, como ya propuso la OMS hace años, como recordó el Panel de Alto Nivel de acceso a medicamentos de la Secretaría General de la ONU en 2016, y como vienen insistiendo diferentes organizaciones y analistas, como Dean Baker, debería financiarse mediante un fondo global, fijando las prioridades de investigación según las necesidades de salud, facilitando licencias abiertas y transferencia de tecnología, propiciando la fabricación en diferentes regiones y garantizando la distribución a precio de coste-plus. Con el sistema actual de monopolios la sociedad paga más de 4 veces todo lo que las empresas dicen que gastan en I+D. Son precios abusivos que los gobiernos no son capaces de controlar, que sangran a los sistemas públicos de salud y a los pacientes, y que provocan, por un lado, falta de acceso a medicamentos necesarios y, por otro lado, sobremedicación y efectos adversos evitables.
La pandemia de la COVID-19 y al acuerdo sobre pandemias de 2025 eran una ocasión para revisar el modelo de forma radical. No se hizo. Los países ricos (presionados por la Big Pharma) impusieron su veto. Tampoco se logró incluir en el acuerdo lo que muchos líderes nacionales defendían en plena pandemia: “Frente a una pandemia se deben suspender los derechos de propiedad intelectual; toda vacuna, tratamiento o medio diagnóstico será accesible a todos, en condiciones de igualdad, a precios de coste, etc.”. Pero cuando se discutió el acuerdo de pandemias habían pasado varios años y la presión social no era tan fuerte. Finalmente, al acuerdo solo incorporó la obligación de que los países (pobres) facilitaran los patógenos con potencial pandémico a la OMS para que pudieran acceder a esta información todas las partes interesadas (países y empresas) y, a cambio, los países y empresas (ricos) se obligaban a garantizar el acceso real a los resultados de posibles investigaciones, vacunas, tratamientos y diagnósticos, a los los países que hubieran facilitado los patógenos. El acuerdo logrado por la OMS en 2025 era un acuerdo cicatero, muy insuficiente. Pero, aun así, los países ricos han estado resistiéndose a asumir estas obligaciones en el anexo PABS por la presión de las grandes empresas farmacéuticas, que no quieren renunciar a sus enormes beneficios, ni siquiera en una pequeña parte. Quieren tener todo el control.
Entristece ver a los líderes de los países europeos dejar de lado los valores que decimos defender, equidad, justicia, solidaridad, y ceder una y otra vez a los intereses de los grandes poderes económicos. Lo paradójico de esta postura es que, en caso de una nueva pandemia de alta contagiosidad y alta letalidad, nadie estará a salvo y la única manera de superar el reto será compartiendo, entre todos y de verdad, el acceso a la información, a los tratamientos y las vacunas. Por eso, desde aquí, insistimos una vez más en que el cambio de modelo en la I+D de medicamentos es cada vez más necesario.
Presidente del Colegio Oficial de Médicos de Toledo.
En junio de 2024, la revista JAMA publicó un estudio (1) basado en un análisis de la base de datos Open Payments de EEUU, base de datos que recoge los pagos y trasferencias de valor de la industria a los profesionales de la salud. Es este estudio se recoge que las compañías farmacéuticas y de dispositivos médicos patrocinaron al menos la friolera de 3.000 eventos diarios dirigidos a los prescriptores, eventos que incluían comidas, honorarios y regalos, y que dichos eventos promocionales se asociaban sin ningún lugar a dudas a un aumento de la prescripción de los productos promocionados, tanto en cantidad, como en coste, con un mayor uso de los medicamentos de las marcas promocionadas sobre los mismos productos en su versión genérica, aunque los propios autores consideran que sus resultados pueden estar claramente infraestimados dado que muchas compañías no están obligadas a presentar informes de estas actividades.
La consistencia de esta relación causa-efecto se ha demostrado una y otra vez consistente en múltiples estudios (2) (3) (4) (5). El siglo XXI nos trajo una preocupación creciente por estas relaciones perversas, y en EEUU comenzaron a aparecer en distintos estados leyes que regulaban estas trasferencias de valor, imponiendo la máxima trasparencia, cristalizadas en la Physician Payments Sunshine Act, conocida como LeySunshine, aprobada por el Senado estadounidense en 2010. En la Unión Europea tuvimos que esperar un año más, al 2011, para que Francia se convirtiera en el primer país de la eurozona en implementar una regulación sobre las relaciones entre médicos e industria farmacéutica, que exigía la divulgación pública de pagos y trasferencias de valor entre ambos.
En nuestro país, parece que debemos conformarnos con un mecanismo autorregulador, el Código de Buenas Prácticas de Farmaindustria, que si bien obliga a las compañías adheridas a la publicación de estos datos, un estudio publicado en la revista Healt Policy en 2021 (6) analizando estos códigos autorreguladores en siete países europeos, entre ellos el nuestro, la realidad es que la trasparencia es limitada y la accesibilidad de los datos es baja, lo que dificulta enormemente un análisis exhaustivo de las relaciones financieras. La gran pregunta sería por qué si las políticas institucionales que restringen las visitas de los representantes farmacéuticos y los regalos han mostrado sin ningún género de duda reducir la prescripción de medicamentos promocionados y aumentar el uso de genéricos, (5) (7) (8) los gobiernos estatales y en nuestro país, los autonómicos, que ostentan las competencias en materia de Sanidad, siguen esquivando su responsabilidad legislativa en este ámbito, ignorando el ahorro en costes que supondría a sus servicios de salud, y confiando como tiernos infantes en que compañías con un lógico ánimo de lucro adopten medidas que les hagan reducir sus cuentas de resultados. Que la respuesta a esta pregunta fuera el que dichos gobiernos y sus servicios de salud obtuvieran de dichas compañías otros beneficios nos provocaría tal ánimo depresivo que sería mejor no planteárnoslo.
Conscientes como espero que seamos todos (al menos los lectores de esta revista) de lo anteriormente expuesto, y tratando de no caer en el desánimo crónico, una plausible línea de actuación que nos permitiera revertir esta corriente de comportamientos con tan discutible componente ético sería fomentar de forma activa, es decir, por el camino de la legislación, la enseñanza sobre la ética de las relaciones con la industria farmacéutica en el periodo del grado y de la formación sanitaria especializada.
Un reciente estudio realizado en Francia mostraba que el 76% de los estudiantes de Medicina nunca habían asistido a una clase sobre las estrategias de marketing que utiliza la industria farmacéutica (9) y el 88% desconocía si su facultad disponla de políticas sobre las interacciones con las farmacéuticas. En Bélgica, solo una de diez facultades de Medicina recogía en los currículums de su profesorado los posibles conflictos de intereses en sus relaciones con la Industria (10). Estudios hechos en EEUU (11) y Japón (12) muestran que la industria muestra interés en ir desarrollando esos vínculos con los futuros prescriptores ya desde su formación académica, y que estas relaciones son percibidas como “naturales” por los estudiantes, que no tienen ningún problema ético en aceptar regalos porque consideran que no les influye de ninguna forma o ya en una época tan precoz de su aprendizaje están instalados en la ilusión de la inmunidad, que seguramente no les abandonará cuando emprendan su actividad profesional. En Europa existen pocos estudios al respecto, aunque en Francia en 2019 (12) se realizó una encuesta nacional entre los estudiantes de Medicina que reveló que el 85% ya había tenido contacto con la Industria en esta etapa, cifras similares a las publicadas sobre los estudiantes alemanes (13) la mayoría de los cuales ya habían participados en eventos promocionados por las farmacéuticas, y cerca de la mitad consideraba que estas relaciones, incluyendo la aceptación directa de regalos, influía más en sus compañeros
que en ellos mismos.
En nuestro país, corrientes como Farmacriticxs, nacida en el seno de IFMSA (Federación española de Asociaciones de Estudiantes de Medicina) en 2009 con la intención de llevar a cabo una reflexión crítica frente a la influencia de la industria en la formación médica, a pesar de sus iniciativas dignas de alabanza (memorable aquellas camisetas con la leyenda “estamos ideando nuevas enfermedades para los nuevos fármacos”) quedan como testimoniales, marginales en una mayoría a la que hemos inculcado que todo aquello que no sea clínica durante su formación es accesorio o directamente fútil.
En resumen, la interacción entre los estudiantes de Medicina y la Industria Farmacéutica es frecuente a nivel global, prácticamente carece por completo de regulación, los estudiantes si bien son capaces de reconocer el potencial de influencia, adquieren rápidamente una sensación de inmunidad que ya no les abandonará y la educación formal sobre los conflictos de intereses es insuficiente o directamente nula.
Una vez expuesta tan cruda realidad, deberíamos pasar a las reflexiones. ¿Damos la batalla por perdida? ¿Es inevitable la contaminación ya desde la Universidad? ¿Seguiremos dando la espalda a una formación reglada, capaz de abordar el problema
en todos sus ángulos?
Se hacen necesarias la imposición de medidas legislativas estatales y autonómicas que obliguen a la inclusión en los planes de estudio de formación obligatoria que aborde sin paños calientes los conflictos de interés, que abran los ojos de los estudiantes a las estrategias de marketing de la industria, que les expongan los conflictos éticos que suponen, la perversión formativa que les puede generar, les arranque de la ilusión de inmunidad, y sobre todo les demuestre que estamos necesitados de un cambio radical, y que dicho cambio vendrá de las nuevas generaciones o seguiremos sumidos en ellas, las mismas arenas movedizas en las que llevamos años hundiéndonos poco a poco.
Referencias
Grundy Q, Held F, MacIsaac M, Baugh CM, Campbell EG, Bero L. Quantifying Industry Spending on Promotional Events Using Open Payments Data. JAMA Health Forum. 2024;5(6):e241581. doi:10.1001/ jamahealthforum.2024.1581 https://jamanetwork.com/journals/jama-health-forum/fullarticle/2820408
Mitchell AP, Trivedi NU, Gennarelli RL, Chimonas S, Tabatabai SM, Goldberg J, Díaz LA Jr, Korenstein D. ¿Están asociados los pagos financieros de la industria farmacéutica con la prescripción médica?: Una revisión sistemática. Ann Pasante Med. 2021 Mar;174(3):353-361. doi: 10.7326/M20-5665. Epub 2020 24 de noviembre. PMID: 33226858; PMCID: PMC8315858. https://pubmed.ncbi.nlm.nih.gov/33226858/
Mitchell AP, Dusetzina SB, Mishra Meza A, Trivedi NU, Bach PB, Winn AN. Pharmaceutical industry payments and delivery of non-recommended and low value cancer drugs: population based cohort study. BMJ. 2023 Oct 25;383:e075512. doi: 10.1136/bmj-2023-075512. PMID: 37879723; PMCID: PMC10599253. https://pubmed.ncbi.nlm.nih.gov/37879723/.
Annapureddy AR, Murugiah K, Zheng L, Minges KE, Grandhi GR, Ross JS, Ahmad T, Rodwin BA, Dhruva SS, Girotra S, Dayoub EJ, Curtis JP, Desai NR. Relationship between industry payments to physicians and prescription patterns for PCSK9is, ARNis and DOACs: A report from the NCDR PINNACLE registry. Am Heart J. 2026 Jan;291:26-36. doi: 10.1016/j.ahj.2025.07.015. Epub 2025 Jul 24. PMID: 40714034; PMCID: PMC12621269. https://pubmed.ncbi.nlm.nih.gov/40714034/
Ansari B. Industry payments and physicians prescriptions: Effect of a payment restriction policy. Soc Sci Med. 2021 Jun;278:113942. doi: 10.1016/j.socscimed.2021.113942. Epub 2021 Apr 16. PMID: 33892242. https://pubmed.ncbi.nlm.nih.gov/33892242/
Mulinari S, Martinon L, Jachiet PA, Ozieranski P. Pharmaceutical industry self-regulation and non-transparency: country and company level analysis of payments to healthcare professionals in seven European countries. Health Policy. 2021 Jul;125(7):915-922. doi: 10.1016/j.healthpol.2021.04.015. Epub 2021 May 4. PMID: 34006392. https://pubmed.ncbi.nlm.nih.gov/34006392/
Larkin I, Ang D, Steinhart J, et al. Association Between Academic Medical Center Pharmaceutical Detailing Policies and Physician Prescribing. JAMA. 2017;317(17):1785–1795. doi:10.1001/ jama.2017.4039 https://jamanetwork.com/journals/jama/fullarticle/2623607
Molina M, Boëffard A, Esvan M, Bastian B. Medical students’ exposure to and attitudes towards product promotion and incentives from the pharmaceutical industry in 2019: a national cross-sectional study in France. BMJ Open. 2022 Jul 20;12(7):e045671. doi: 10.1136/bmjopen-2020-045671. PMID: 35858728; PMCID: PMC9305804. https://pubmed.ncbi.nlm.nih.gov/35858728/
Bechoux L, De Vleeschouwer O, Vanheuverzwijn C, Verhegghen F, Detiffe A, Colle F, Fallon C, Thoreau F. Conflict of interest policies at Belgian medical faculties: Cross-sectional study indicates Little oversight. PLoS One. 2021 Feb 10;16(2):e0245736. doi: 10.1371/journal.pone.0245736. PMID: 33566836; PMCID: PMC7875358. https://pubmed.ncbi.nlm.nih.gov/33566836/
Sierles FS, Brodkey AC, Cleary LM, et al. Medical Students’ Exposure to and Attitudes About Drug Company Interactions: A National Survey. JAMA. 2005;294(9):1034–1042. doi:10.1001/jama.294.9.1034 https://jamanetwork.com/journals/jama/fullarticle/201475
Saito S, Maeno T, Miyata Y, Maeno T. Medical students’ attitudes toward interactions with the pharmaceutical industry: a national survey in Japan. BMC Med Educ. 2018 Dec 4;18(1):286. doi: 10 . 1186 / s12909 – 018 – 1394 – 9 . PMI D: 30509273 ; PMCID: PMC 627817. https://pubmed.ncbi.nlm.nih.gov/30509273/
Molina M, Boëffard A, Esvan M, Bastian B. Medical students’ exposure to and attitudes towards product promotion and incentives from the pharmaceutical industry in 2019: a national cross-sectional study in France. BMJ Open. 2022 Jul 20;12(7):e045671. doi: 10.1136/bmjopen-2020-045671. PMID: 35858728; PMCID: PMC9305804. https://pubmed.ncbi.nlm.nih.gov/35858728/
Lieb K, Koch C. Medical students’ attitudes to and contact with the pharmaceutical industry: a survey at eight German university hospitals. Dtsch Arztebl Int. 2013 Sep;110(35-36):584-90. doi: 10.3238/arztebl.2013.0584. Epub 2013 Sep 2. PMID: 24078838; PMCID: PMC3785017. https://pubmed.ncbi.nlm.nih.gov/24078838/
Farmacéutico, coautor junto a su hermano Raúl del ensayo “De venta de farmacias. Una denuncia del negocio de la salud desde dentro”.
El pasado mes de octubre de 2025 tuve el placer de asistir, junto con mi compañero Asier, al XVI Encuentro Global de Parlamentarios de Sanidad organizado por el diario especializado Redacción Médica. El placer no fue pleno, ya que se nos invitó a asistir como público y no como participantes en ninguna mesa de debate. Una decisión comprensible si se considera que ni Asier ni yo somos parlamentarios, pero que aun así nos dejó un cierto “regustillo” de oportunidad perdida: la sensación de que podríamos haber aportado una visión más disruptiva de la sanidad, menos condicionada por inercias partidistas y más abierta a repensar los marcos conceptuales sobre los que se construye la política sanitaria.
Entre las distintas mesas organizadas, me llamó especialmente la atención la dedicada a la innovación terapéutica y al acceso equitativo a terapias avanzadas. Bajo el título “El acceso a terapias innovadoras en el SNS”, la mesa reunió a responsables políticos de varias comunidades autónomas y representantes de diversos partidos. La mayoría eran médicos, lo cual aportó sensibilidad clínica, pero también dejó entrever ciertos sesgos del propio debate: una visión centrada en la experiencia asistencial, pero quizá menos atenta a los determinantes estructurales, económicos y regulatorios que condicionan la incorporación de medicamentos al sistema sanitario.
El eje de la discusión fue, mayoritariamente, el tiempo de acceso a las nuevas terapias. Los ponentes destacaban que, según los últimos datos, los plazos de disponibilidad se han reducido hasta los 616 días en 2024, aunque aún se mantienen por encima de los 518 días de la media europea. Esta afirmación se repetía como mantra: España debe ser más rápida, más ágil, menos burocrática. Una lectura comprensible si se parte de la premisa, no siempre explicitada, de que todo medicamento autorizado supone un avance clínico sustancial que debería llegar cuanto antes a los pacientes. De ahí la insistencia en acortar los procedimientos nacionales tras la autorización regulatoria.
Otro aspecto muy subrayado fue el elevado precio de muchas terapias recientes. Se planteó la necesidad de centralizar los pagos a nivel estatal, en lugar de fragmentarlos por comunidades autónomas, para evitar desigualdades territoriales. También se mencionó la posibilidad de fraccionar pagos o buscar modelos alternativos como abonos plurianuales. Estas propuestas, razonables en un contexto de creciente presión presupuestaria, parten de una constatación: muchos de los medicamentos presentados como innovadores tienen precios que superan varias decenas de miles de euros por paciente y año. Ante ello, los ponentes insistieron en la idea de que, si la Agencia Europea del Medicamento (EMA) ha dado su visto bueno, el resto del proceso debería ser casi automático. “Más velocidad y menos burocracia”, se llegó a comentar.
Sin embargo, en todo ese debate faltó una pregunta esencial: ¿qué es realmente una innovación terapéutica? Y, en consecuencia, ¿todo lo que aprueba la EMA puede considerarse una innovación digna de financiación prioritaria? Es significativo que nadie planteara esta cuestión en una mesa que discutía precisamente el acceso a la innovación. Porque, sin una definición clara de qué entendemos por innovación terapéutica, es difícil evaluar si estamos acelerando lo que merece ser acelerado o simplemente reproduciendo inercias que benefician más a la industria que al interés público.
Responder a esta pregunta implica abandonar la idea de que innovación es sinónimo de novedad. Un mecanismo de acción distinto, una formulación más moderna o una nueva indicación no constituyen, por sí mismos, una innovación clínica relevante. Desde una perspectiva de salud pública y de evaluación de tecnologías, una innovación terapéutica solo puede considerarse como tal si aporta un valor incremental significativo respecto a las alternativas disponibles. Ese valor puede manifestarse de múltiples maneras: una mejora sustancial en variables duras como la mortalidad o los eventos mayores; una mejora relevante en la calidad de vida evaluada con instrumentos validados; una reducción clara de efectos adversos graves; una simplificación del tratamiento con impacte real en adherencia y pronóstico; o la cobertura de una necesidad terapéutica no resuelta por ninguna otra intervención disponible. Ninguno de estos criterios tiene que ver con la novedad comercial o con la rapidez en la aprobación. Tienen que ver con resultados tangibles para los pacientes.
Por el contrario, no deberían considerarse innovaciones terapéuticas los medicamentos que logran simplemente demostrar no inferioridad frente a comparadores subóptimos, o que se apoyan en variables subrogadas sin correlación demostrada con desenlaces clínicos reales, o que carecen de estudios comparativos frente a la mejor alternativa disponible. La literatura internacional es clara: entre el 70% y el 85% de los nuevos fármacos que salen al mercado no ofrecen un beneficio clínico adicional significativo. Son nuevos, pero no mejores; a menudo más caros, pero no más eficaces; y presentados como avances disruptivos, pero sin traducirse en mejoras reales en la salud.
Esto nos lleva inevitablemente al papel de la EMA. Conviene recordar que se trata, prácticamente, de una agencia autorizadora, no de una agencia de evaluación de valor. Su función es determinar si un medicamento es eficaz y seguro en relación con los ensayos aportados por su promotor, y lo hace comparando frecuentemente con placebo o con tratamientos que no necesariamente representan el estándar óptimo. La EMA no evalúa ni el coste-efectividad, ni el valor incremental, ni el impacto presupuestario, ni la pertinencia clínica en el contexto nacional. Su aprobación es un requisito regulatorio imprescindible, pero no equivale a un aval de innovación terapéutica. A ello se añade un elemento que debería generar, como mínimo, reflexión institucional: más del 90% de su financiación procede de tasas abonadas por las compañías farmacéuticas por la evaluación y mantenimiento de autorizaciones. Esto no implica, por sí mismo, un conflicto de interés directo, pero sí configura un modelo donde la estructura regulatoria depende económicamente de quienes buscan la aprobación de sus productos. En ese contexto, es imprescindible que los Estados miembro cuenten con mecanismos sólidos de evaluación del valor clínico real que sirvan de contrapeso.
La importancia de estos matices se hace evidente al revisar algunos ejemplos recientes en los que la narrativa comercial de la innovación no se corresponde con su valor terapéutico incremental. Lebrikizumab (Ebglyss®), autorizado para dermatitis atópica moderada-grave y comercializado a más de 1.200€ por jeringa, fue evaluado en Alemania sin que se demostrara un beneficio adicional claro frente al comparador apropiado, dupilumab. La ausencia de superioridad clínica convierte esta supuesta innovación en un ejemplo de novedad molecular sin verdadero avance. Algo similar ocurre con Zilucoplan (Zilbrysq®), indicado para miastenia gravis generalizada y cuyo coste semanal oscila entre 3.400€ y 6.600€. Las evaluaciones independientes concluyeron, nuevamente, que no existía un valor añadido probado. Bimekizumab (Bimzelx®), utilizado en patologías inflamatorias y presentado como una molécula con un doble mecanismo de acción sobre IL-17A e IL-17F, tampoco mostró un beneficio incremental significativo frente a las alternativas ya disponibles, pese a su elevado precio por pluma. Por último, Inebilizumab (Uplizna®), destinado a la neuromielitis óptica y con un precio que supera los 8.000€ por vial, no logró demostrar superioridad frente a rituximab ni mejoras relevantes en desenlaces clínicos duros. Su mecanismo dirigido a CD19 puede ser novedoso, pero la novedad biológica no implica necesariamente un avance terapéutico real.
Antes de continuar, conviene aclarar un aspecto fundamental del debate sobre precios. A menudo se citan cifras extraídas de los acuerdos de la Comisión Interministerial de Precios de Medicamentos (CIPM), pero estos valores representan únicamente el precio industrial máximo. En la práctica, el coste real que afronta el Sistema Nacional de Salud puede ser (y suele ser) sustancialmente distinto gracias a descuentos confidenciales, techos de gasto, pagos por resultados y acuerdos de riesgo compartido. Esta opacidad, común en toda Europa, tiene una implicación directa para el debate sobre innovación: el precio de lista no es un indicador fiable del valor real. Un fármaco puede tener un precio nominal altísimo, pero un coste efectivo muy inferior, o al contrario. Lo relevante no es cuánto figura en la ficha financiera, sino cuánto paga realmente el sistema y qué valor clínico obtiene a cambio.
Estos ejemplos muestran algo crucial: eficacia no es sinónimo de innovación. Un medicamento puede ser eficaz (la mayoría dicen serlo) y, aun así, no aportar una mejora sustancial frente a las terapias disponibles. Del mismo modo, un precio elevado no implica automáticamente mayor valor clínico. La aceleración indiscriminada en la incorporación de nuevos medicamentos, si no va acompañada de una reflexión rigurosa sobre su valor incremental, puede comprometer la sostenibilidad del sistema, desplazar recursos hacia tratamientos menos eficientes e incluso generar inequidades territoriales. La disyuntiva real no es burocracia frente a velocidad, sino valor frente a coste.
Ahora bien, el escepticismo razonado no debe confundirse con rechazo a la verdadera innovación. Los últimos años han traído avances que sí han cambiado la práctica clínica de forma profunda: las terapias CAR-T, determinadas terapias avanzadas para enfermedades raras, inhibidores dirigidos con impacto demostrado en supervivencia global y medicamentos capaces de transformar enfermedades letales en patologías crónicas controlables. Estos tratamientos sí cumplen los criterios de innovación terapéutica: aportan beneficios clínicos sustanciales, duraderos y relevantes. Precisamente por eso debemos proteger la capacidad del Sistema Nacional de Salud para financiarlos. Si todo se etiqueta como avance, nada lo es. Si dedicamos recursos a terapias sin valor incremental, la consecuencia paradójica es que dificultamos la incorporación de las verdaderas innovaciones.
Por ello, las decisiones políticas en materia de medicamentos no pueden reducirse a un debate sobre rapidez. Deben situarse en el terreno de la estrategia, de la planificación a largo plazo y de la claridad conceptual. La innovación terapéutica debe entenderse como un bien público cuya financiación exige criterios rigurosos de valor clínico y coste-efectividad, no como un producto tecnológico cuyo precio viene determinado por la narrativa comercial. La responsabilidad política consiste en garantizar que cada euro invertido en medicamentos genera el máximo beneficio social posible. Y eso implica decidir, no simplemente gestionar; orientar la innovación, no dejarse arrastrar por ella; priorizar las terapias que realmente transforman la vida de los pacientes, no aquellas que solo transforman los balances de las compañías.
Solo desde esta perspectiva (más exigente, más transparente y más orientada al valor) el Sistema Nacional de Salud podrá conciliar innovación, equidad y sostenibilidad. Solo así podrá seguir siendo un motor de acceso justo, un garante de racionalidad y un actor capaz de orientar, y no simplemente seguir, la evolución futura de la innovación farmacéutica.
Médico Interno Residente de Medicina Preventiva y Salud Pública en el Hospital Clínico San Carlos (Madrid). Graduado en Medicina por la Universidad Complutense de Madrid. Vocal de la Asociación por el Acceso Justo al Medicamento (AAJM).
El pasado jueves 12 de febrero de 2026 tuvo lugar en la sede de la Fundación de Ciencias del Medicamento y Productos Sanitarios (Fundamed) el Encuentro de Saberes “Aspectos sociales del medicamento” con motivo de la creación de la cátedra Bidafarma-Universidad Rey Juan Carlos. En este evento tuvo lugar la mesa titulada “El medicamento como determinante de salud en una sociedad cambiante”, a la cual fue invitada la Asociación de Acceso Justo al Medicamento (AAJM). En la mesa también participaron Javier Vinzia Portabella, Director de Banco Farmacéutico; Jara Zotes Ciprés, del Observatorio de la pobreza farmacéutica; Núria Llurba Montesinos, de Farmacéuticos Mundi (Farmamundi); y un servidor.
Durante esta mesa pude hacer una exposición de los siguientes puntos:
1. EL medicamento como determinante de salud
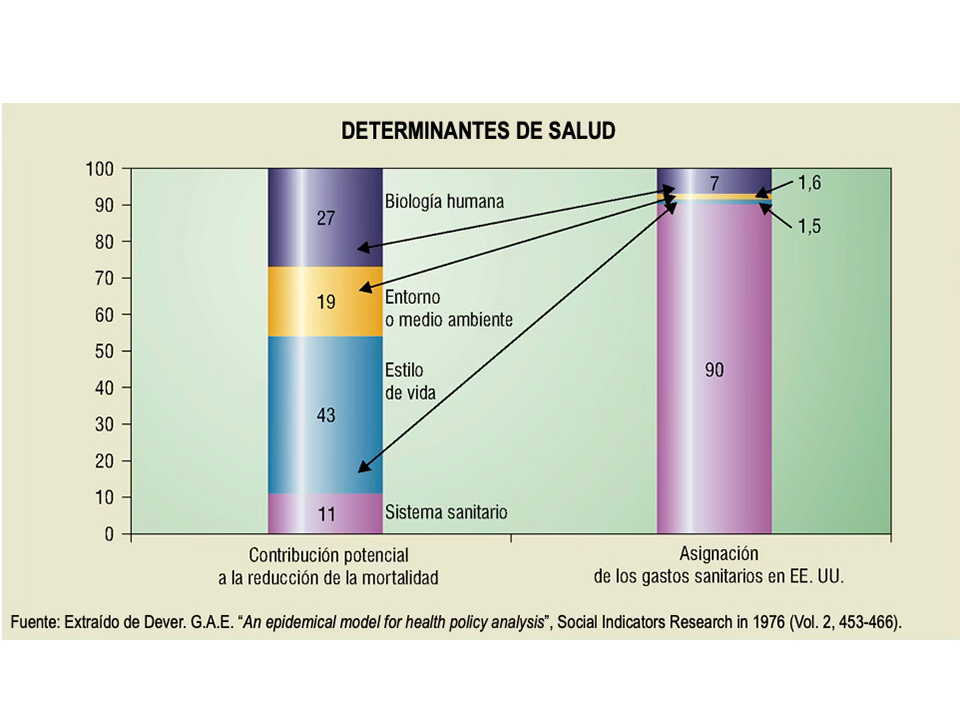
Durante la inauguración, por parte del Catedrático de Medicina Preventiva y Salud Pública, Ángel Gil de Miguel, se comentó «A New Perspective on the Health of Canadians» (1). Es un informe elaborado en 1974 por el Ministerio de Salud y Bienestar del Gobierno de Canadá que se convirtió en una referencia en las políticas de salud pública y promoción de la salud. Conocido mundialmente como Informe Lalonde, en honor al Ministro de Salud canadiense, el documento propone la fórmula para integrar los sistemas sanitarios y la promoción de la salud en el marco de una política integral. En este primer documento describe 4 determinantes de salud:
Estilos de vida: Decisiones personales (dieta, ejercicio, adicciones) que impactan en la salud.
Medio ambiente: Factores físicos, sociales y culturales (contaminación, entorno socioeconómico).
Biología humana: Aspectos genéticos, hereditarios y envejecimiento.
Organización de la atención sanitaria: Calidad, accesibilidad y cobertura de los servicios de salud.
Contrasta entre estos 4 determinantes, cómo pese a que la organización de la atención sanitaria representa un porcentaje pequeño en la salud de las personas, se lleva un gran porcentaje del presupuesto anualmente.
Este documento creó un marco que fue ampliado por Dahlgren y Whitehead en 1991 con sus “Determinantes sociales de la salud” (2).
En este marco, el medicamento forma parte del sistema sanitario, pero está fuertemente atravesado por los determinantes sociales, porque para acceder a él se ha necesitado 1) identificar la necesidad en salud, y para ello es necesaria un alfabetismo sanitario y unas creencias, confianza y expectativas en el sistema sanitario; 2) capacidad para la búsqueda de asistencia sanitaria, determinada por los valores sociales, culturales, de género, autonomía; 3) capacidad para alcanzar los servicios sanitarios, determinada por el transporte y la movilidad, el entorno, el soporte social, así como la localización y accesibilidad geográfica de los servicios, las horas de apertura, los mecanismos para las citas; 4) la capacidad de financiarlo, en caso de que haya copagos, o sean prestaciones que no estén en la cartera, depende también de los costes de oportunidad (cuánto dinero está perdiendo dejando de ir al trabajo para ir a la consulta); 5) la capacidad para continuar con el proceso, que depende de la información que reciba, de la adherencia, del empoderamiento, del soporte de los cuidadores, etc. (3)
2. Derecho a la salud y asistencia médica
El derecho a la salud, del que se deriva, en los sistemas sanitarios modernos, el acceso efectivo a medicamentos esenciales, está recogido en la Declaración de los Derechos Humanos en el artículo 25 (4), en nuestra Constitución Española como principio rector en el artículo 43, así como la competencia de los poderes públicos para organizar y tutelar las prestaciones sanitarias (5) y en la Ley General de Sanidad, se dice “Todos tienen los siguientes derechos con respecto a las distintas administraciones públicas sanitarias:” entre los que se incluye en el punto 14. “A obtener los medicamentos y productos sanitarios que se consideren necesarios para promover, conservar o restablecer su salud, en los términos que reglamentariamente se establezcan por la Administración del Estado.” (6)
No obstante, en el barómetro sanitario de 2025, en la pregunta 13 “En los últimos doce meses, ¿ha dejado Ud. de tomar algún medicamento recetado por un/a médico/a de la sanidad pública porque no se lo pudo permitir por motivos económicos?”, un 4,6% declaraba que no tomaba su medicación prescrita por motivos económicos, lo que, extrapolado a la población española, corresponde a 2,2 millones de personas para las cuales, el precio del medicamento es una barrera.
Esta barrera económica, tiene un nombre: copago. El Real Decreto-ley 16/2012, de medidas urgentes para garantizar la sostenibilidad del Sistema Nacional de Salud y mejorar la calidad y seguridad de sus prestaciones, a parte de expulsar a las personas migrantes sin papeles, pasando de una semi-universalidad a recibir prestaciones en función de la condición de trabajador, también expulsa de facto a aquellas personas que por motivos económicos no pueden permitirse el copago farmacéutico.
Fueron interesantes las aportaciones de Javier Vinzia Portabella, Director de Banco Farmacéutico y Jara Zotes Ciprés, del Observatorio de la pobreza farmacéutica, al aportar datos: el 91,2% de los beneficiarios del banco farmacéutico, pertenecen al grupo TSI 003, es decir, trabajadores activos con una renta inferior a 18.000 euros anuales, a quienes corresponde una aportación farmacéutica del 40% del precio del medicamento.
3. Sostenibilidad
La población ha cambiado. Gracias a las mejoras de las condiciones de vida (educación general y sanitaria, derechos laborales, protección del medio ambiente, mejora de la nutrición, etc.) podemos disfrutar de una población más longeva, pero con más enfermedades crónicas. Nos enfrentamos a un reto de más población que además tiene más necesidades objetivas.
Pero además, tenemos un proceso de medicalización que genera aún más necesidades:
Avances diagnósticos y terapéuticos: para quien tiene un martillo, todo son clavos. Cada año crece la lista de diagnósticos posibles. También, más malestares son susceptibles de ser medicalizados, especialmente pero no solo, en el ámbito de la salud mental.
Mayor demanda: la salud es un bien de mercado inagotable porque siempre podemos estar mejor.
Interés industrial-comercial: claramente existen intereses comerciales y toda una industria farmacéutica que va a querer aumentar la prescripción. Hablamos de sus estrategias más adelante.
Todo esto lleva a que el gasto público en farmacia aumente año tras año. Pero no solo se debe a un aumento de la demanda, también a un aumento del precio.
4. Determinantes comerciales de la salud
Si antes hemos hablado de los determinantes sociales, hemos de hablar de los determinantes comerciales de la salud. Si bien las entidades comerciales pueden contribuir positivamente a la salud y la sociedad, existe una creciente evidencia de que los productos y prácticas de algunos actores comerciales- en particular las corporaciones transnacionales más grandes son responsables de las crecientes tasas de mala salud evitable, daño planetario e inequidad social y de salud; estos problemas se conocen cada vez más como los determinantes comerciales de la salud. (7)
Habitamos el capitalismo. Estamos en un sistema económico donde una proporción sustancial de los medios de producción y propiedad está operada por particulares para obtener una ganancia. Por otra parte, tenemos unas necesidades: producción y abastecimiento de medicamentos, investigación y desarrollo de nuevas terapias.
Dado que estamos en un sistema financiado fundamentalmente por las arcas públicas, esto hace que el sistema sea susceptible de sufrir captura corporativa (10), mediante la cual, las decisiones políticas responden a intereses privados. Esta captura corporativa tiene una parte material: donaciones a partidos, puertas giratorias o incluso actos ilegales como sobornos; social, mediante el marketing, promoción de enfermedades, financiación de organizaciones y plataformas de pacientes; e intelectual, influyendo en la formación, tanto de grado financiando cátedras, como de posgrado financiando la formación continuada, congresos y sociedades científicas, controlan las prioridades de la investigación, la publicación a través de las revistas científicas y la recomendación con las guías clínicas.
5. Capitalismo sin libre mercado: la patente
El libre mercado es un sistema económico en el cual los precios de los bienes y servicios son determinados por la interacción de intercambios voluntarios entre los individuos según la ley de la oferta y la demanda. Requiere para su funcionamiento la existencia de la libre competencia y precios libres y para ello es necesario el respeto por los derechos de propiedad y de contrato entre las partes, en ausencia de coacción para la toma de decisiones. En economía, un fallo de mercado es una situación en la que la asignación de los recursos por parte del mercado no es eficiente.
Llamamos “Competencia imperfecta” cuando una empresa tiene más poder de mercado que el resto de las empresas que están operando en un momento determinado. Como consecuencia de este fallo, los consumidores van a consumir una cantidad menor a un precio mayor. Un claro ejemplo de estructura de mercado sin desempeño óptimo de acuerdo a los patrones de eficiencia económica son es el monopolio. En el caso del medicamento, este monopolio tiene un soporte legal llamado patente.
En España, el Real decreto-ley de 26 de Julio de 1929 sobre Propiedad Industrial (11), documento que regulaba las patentes, indicaba explícitamente en su artículo 48 que “No podrán ser objeto de patente: 2.° Los productos o los resultados industriales, las fórmulas farmacéuticas y medicamentosas y las de los alimentos para la especie humana o los animales; pero sí lo serán los procedimientos y los aparatos para obtenerlos.” No obstante, cuando España firma el Acuerdo sobre los Aspectos de los Derechos de Propiedad Intelectual relacionados con el Comercio (ADPIC) (12) en 1994, esta excepción se elimina, otorgando un monopolio al patentador durante 20 años. Este monopolio confiere poder de mercado que permite imponer precios elevados, con sobrecostes y opacidad, y una investigación fuertemente condicionada por la rentabilidad. Aunque el ADPIC contempla flexibilidades como las licencias obligatorias y voluntarias, su uso en la práctica es excepcional en países de renta alta.
6. El valor del medicamento
Es fundamental que diferenciemos entre valor y precio. El medicamento tiene un valor incalculable, porque en ocasiones separa la vida de la muerte. Pero el valor no puede ser un argumento para maximizar los beneficios de la industria. Por analogía: una apendicectomía intervenida en la privada tiene un precio de entre 3000 y 7000€. Tiene un gran valor social porque no se muere una persona de 17 años, que además va a estar cotizando durante toda su vida. ¿Por qué no cuesta más? Porque no tiene sentido que el precio venga determinado por el valor.
Como mensajes clave de la exposición, quise transmitir:
Que la industria privada siempre va a tener lucro como fin, a costa de las arcas públicas y las necesidades de la población.
Que es necesario tomar medidas como:
Centralización de compras
Publicación de costes de I+D
Fin de la confidencialidad en precios
Eliminación de copagos o reforma según renta real
Que con el ahorro conseguido cuando se eviten los sobrecostes se ha de financiar la independencia de la formación de los profesionales y de la labor de las organizaciones civiles.
Levesque JF, Harris MF, Russell G. Patient-centred access to health care: conceptualising access at the interface of health systems and populations. International Journal for Equity in Health. 2013; 12(18). DOI: https://doi.org/10.1186/1475-9276-12-18
Jefatura del Estado. Real Decreto-ley 16/2012, de 20 de abril, de medidas urgentes para garantizar la sostenibilidad del Sistema Nacional de Salud y mejorar la calidad y seguridad de sus prestaciones. Disponible en: https://www.boe.es/buscar/act.php?id=BOE-A-2012-5403
Gilmore AB, Fabbri A, Baum F, et al. Definición y conceptualización de los determinantes comerciales de la salud. The Lancet. 2023; 401(10383): 1194-1213. DOI: https://doi.org/10.1016/S0140-6736(23)00013-2
Royo-Bordonada MA. Captura corporativa de la salud pública. Rev. Bioética y Derecho. 2019;(45):25-41. ISSN 1886-5887
A continuación, recogemos los comentarios realizados por expertos bien reconocidos sobre el documento, firmado por Public Citizen, Health Gap y otras organizaciones, donde se expone la necesidad de resistir a los ataques del gobierno Trump que cuestionan un acceso justo y asequible a los medicamentos.
Este texto es realmente una llamada a la acción mundial para impedir que las medidas arancelarias y de política farmacéutica de Trump provoquen una situación desastrosa en el acceso de la vacunas y medicamentos necesarios para preservar, y mantener la salud de la humanidad.
(Finalizado los comentarios, el lector/a puede encontrar el texto completo y las organizaciones firmantes).
WASHINGTON, D.C. — Public Citizen, Health GAP y 99 organizaciones expertas de todo el mundo exigen un enfoque de política comercial global que preserve el acceso a medicamentos asequibles y rechace los acuerdos alcanzados bajo presión.
El acuerdo de principio entre EEUU. y el Reino Unido con las grandes farmacéuticas, fruto de un abuso de poder comercial y aranceles instrumentalizados, no debe repetirse. Aun así, en las semanas posteriores al acuerdo con el Reino Unido, EEUU. obligó a Argentina a firmar un Acuerdo de Comercio Recíproco (TAR) que impulsa la agenda monopolística de las grandes farmacéuticas a expensas de la salud pública. Expertos de los sectores de salud pública, comercio, trabajo, clima y religión insisten en destacar que en los enfoques comerciales se debe preservar la capacidad de los países para:
Garantizar precios asequibles para todos
Rechazar la intimidación corporativa
Permitir un suministro abundante de medicamentos
Garantizar la seguridad, eficacia y calidad de los medicamentos
Determinar libremente qué tratados internacionales son beneficiosos
Adherirse a procesos comerciales transparentes
El director de Acceso a Medicamentos de Public Citizen, Peter Maybarduk, emitió la siguiente declaración:
“Las amenazas arancelarias de Trump solo conducen a un agravamiento de la escasez y el racionamiento de medicamentos. Su alianza deshonesta con las grandes farmacéuticas para aumentar los precios de los medicamentos en el extranjero pone en riesgo la vida de las personas, a la vez que distrae del cambio real y la rendición de cuentas de la industria farmacéutica que necesitamos en Estados Unidos para que los medicamentos sean asequibles.
En lugar de aumentar los costos y lanzar amenazas, los gobiernos deberían invertir para expandir la producción de medicamentos en todas partes y aprender de las mejores prácticas de los demás para reducir los precios en sus países”.
Brook Baker, asesor principal de políticas de Health GAP, emitió la siguiente declaración:
“La administración Trump ha diezmado la programación y la prestación de servicios de salud a nivel mundial mediante reducciones y restricciones en la ayuda exterior para la salud, pero también busca consolidar el poder monopolístico de las grandes farmacéuticas mediante acuerdos comerciales apresurados y negociados en secreto, y amenazas comerciales. En lugar de fomentar suministros adecuados, precios asequibles y un acceso equitativo a las tecnologías médicas, Trump aplica normas regulatorias más rigurosas en materia de propiedad intelectual, protección de precios y que maximizan las ganancias de las farmacéuticas a expensas de la salud, intimidando a los países con aranceles y otras medidas. Los seis principios para el comercio y el acceso a los medicamentos contrarrestan la hegemonía de las farmacéuticas y las medidas coercitivas de Trump en su nombre”.
El investigador de Acción Internacional para la Salud, Javier Llamozaemitió el siguiente comunicado:
«Latinoamérica tiene el derecho soberano de regular, producir y adquirir medicamentos seguros y rentables, priorizando la vida por encima del lucro. Rechazamos acuerdos que refuercen monopolios, encarezcan los tratamientos y debiliten nuestras capacidades locales. La salud no se negocia bajo presión ni amenazas comerciales».
«América Latina tiene el derecho soberano de regular, producir y adquirir medicamentos seguros y asequibles, priorizando la vida sobre las ganancias. Rechazamos acuerdos que fortalecen los monopolios, aumentan el costo de los tratamientos y debilitan nuestras capacidades locales. La salud no es negociable bajo presiones o amenazas comerciales».
Tim Bierley, director de Políticas y Campañas de Global Justice Now, emitió la siguiente declaración:
“Durante años, el Reino Unido ha aplicado controles de precios sensatos para limitar el impacto de los notorios sobreprecios de las farmacéuticas por los medicamentos. Al ceder en las negociaciones con Trump y la industria farmacéutica y aceptar suavizarlos, nuestro gobierno ha comprometido al Reino Unido con una atención médica más cara sin nada tangible a cambio.
Este terrible acuerdo también sienta un precedente preocupante, incentivando a las farmacéuticas a usar tácticas de extorsión para aumentar los precios de los medicamentos en todo el mundo. En lugar de actuar en solitario, los países deben trabajar juntos para combatir el poder monopolístico de las farmacéuticas, que siempre priorizarán la ganancia sobre nuestra salud”.
Sangeeta Shashikant, asesora legal y de políticas de la Red del Tercer Mundo, emitió la siguiente declaración:
“Los gobiernos tienen el deber legal y moral de garantizar el derecho a la salud de sus ciudadanos. La política comercial nunca debe utilizarse como arma para socavar esa responsabilidad ni para obligar a los países a desmantelar sus salvaguardias de salud pública. Cualquier marco comercial que restrinja el margen de maniobra, debilite la producción local o limite el acceso a medicamentos asequibles es fundamentalmente incompatible con los derechos humanos, especialmente el derecho a la salud, y debe ser rechazado”
Principios para el Acceso a los Medicamentos y el Comercio (texto completo)
La administración Trump está utilizando el poder comercial de Estados Unidos y aranceles extremos para intimidar a otros países y obligarlos a firmar acuerdos vinculantes que socavan el acceso asequible y fácil a los medicamentos.
El caos comercial de Trump exige precios más altos para los medicamentos fuera de Estados Unidos, antepone los intereses corporativos a las necesidades sanitarias, promueve aranceles farmacéuticos elevados que reducen el suministro mundial, socava el desarrollo de la capacidad local y persigue todas sus demandas mediante negociaciones secretas.
Los países que se apresuran a satisfacer las exigencias de Estados Unidos corren el riesgo de aceptar condiciones —incluidas las disposiciones perjudiciales en materia de propiedad intelectual y restricciones a la negociación, la adquisición y la producción de medicamentos— que comprometen el acceso a los medicamentos y el derecho a la salud.
Creemos que los enfoques comerciales deben preservar la capacidad de los países para:
1. Garantizar precios asequibles para todos.
2. Rechazar el acoso corporativo.
3. Permitir un suministro abundante de medicamentos.
4. Garantizar la seguridad, la eficacia y la calidad de los medicamentos.
5. Determinar libremente qué tratados internacionales son beneficiosos.
6. Adherirse a procesos comerciales transparentes y responsables.
Garantizar precios asequibles para todos.
Trump quiere que las empresas farmacéuticas aumenten sus precios en muchos países, lo que podría provocar el racionamiento de los tratamientos y agotar los presupuestos sanitarios. Su excusa: es que esto, de alguna manera, reducirá los precios para los estadounidenses. Pero no hay motivos para pensar que unos precios más altos en algunos países provocarán una bajada de los precios, o la aplicación de la cláusula de «nación más favorecida», en Estados Unidos. Por el contrario, esta política amenaza con incrementar los precios ya elevados, al dar a las empresas con poder monopolístico más libertad para fijar precios excesivos.
Los medicamentos deben ser asequibles para los particulares y los pagadores públicos y privados, y sostenibles para los sistemas de salud.
Para conseguir precios más justos, hay que evitar las medidas que buscan desregular los precios de los productos farmacéuticos.
Muchos países negocian los precios y evalúan la eficacia clínica y la rentabilidad para garantizar la asequibilidad de los nuevos medicamentos. El Reino Unido utiliza estos sistemas para contrarrestar los precios excesivamente elevados fijados por las empresas farmacéuticas y mitigar el impacto presupuestario.
Sin embargo, como resultado de la presión ejercida por la administración Trump y la industria farmacéutica, el Reino Unido ha aceptado modificar sus normas de fijación de precios de los medicamentos, lo que se traducirá en un aumento del gasto farmacéutico. Además, algunas empresas farmacéuticas ya han aceptado aumentar los precios de entrada en el Reino Unido.
2. Rechazar el acoso corporativo.
Si bien la presión sostenida obligó a las normas comerciales internacionales a reconocer la importancia de salvaguardar la salud pública y el acceso a los medicamentos, los acuerdos comerciales suelen incluir cláusulas de propiedad intelectual que van más allá de las normas acordadas internacionalmente, entre otras cosas al exigir normas menos estrictas de patentabilidad, exclusividad de datos/comercialización (monopolios), prórrogas de la vigencia de las patentes y vinculación entre la patente y la situación reglamentaria. A menudo se utilizan amenazas comerciales y políticas para limitar el uso de las licencias obligatorias. Esto ha reducido el margen de maniobra de los países para facilitar el acceso a medicamentos genéricos asequibles y combatir las prácticas anticompetitivas. Además, Estados Unidos suele buscar una mayor protección de los secretos comerciales en los acuerdos comerciales, lo que permite a las empresas mantener la confidencialidad de información importante y esconderse tras
acuerdos de no divulgación.
Los países deben tener libertad para promulgar y aplicar políticas que permitan el acceso a los medicamentos sin restricciones derivadas de las normas comerciales ni presiones externas.
Las presiones comerciales nunca deben utilizarse para socavar o intentar eliminar el margen de maniobra acordado internacionalmente en materia de salud pública y propiedad intelectual. Los gobiernos no deben fomentar la adopción ni aceptar compromisos en materia de propiedad intelectual en los acuerdos comerciales que limiten la capacidad de proteger o aumentar el acceso a medicamentos asequibles. Los países deben seguir siendo libres de exigir la divulgación pública de los costos de investigación y desarrollo y de fabricación, los precios, los acuerdos de suministro y otra información farmacéutica de interés público.
Los gobiernos tienen derecho a aplicar salvaguardias de interés público en sus leyes y
prácticas en materia de propiedad intelectual. La Ley de Patentes de la India, por ejemplo, incluye importantes salvaguardias que favorecen el acceso asequible a los medicamentos al impedir la concesión de patentes de baja calidad que prolongan los monopolios sobre los medicamentos.
El Relator Especial de las Naciones Unidas sobre el derecho a la salud, reconociendo el impacto adverso que el Acuerdo sobre los ADPIC de la OMC y los acuerdos de libre comercio han tenido en los precios y la disponibilidad de los medicamentos, recomienda incorporar flexibilidades como estas en las leyes nacionales para facilitar el acceso a los medicamentos genéricos. El Relator Especial recomienda, además que los países en desarrollo no introduzcan normas ADPIC-plus en sus leyes nacionales y que los países desarrollados no alienten a los países en desarrollo a celebrar acuerdos comerciales ADPIC-plus.
3. Permitir un suministro abundante de medicamentos.
Tras la grave escasez de vacunas y tratamientos contra la COVID-19, existe un consenso cada vez mayor en torno a la necesidad de reforzar las capacidades biofarmacéuticas locales y regionales para respaldar, la respuesta de emergencia, atender las necesidades sanitarias locales, garantizar un suministro oportuno y adecuado, y promover la ciencia y la investigación a nivel mundial. Sin embargo, los enfoques comerciales pueden ir en contra de estos objetivos.
El suministro oportuno, asequible y equitativo de medicamentos requiere la participación de una gran variedad de productores de todo el mundo.
Fomentar una mayor capacidad de producción e investigación, especialmente en los países de ingresos bajos y medios, entre otras cosas facilitando el intercambio de la propiedad intelectual, la tecnología y los conocimientos necesarios para producir instrumentos médicos.
Las políticas comerciales destinadas a respaldar las políticas industriales deben estar en
consonancia con los objetivos sanitarios y apoyar el acceso sostenible a los medicamentos en todas partes.
Las iniciativas para aumentar la producción nacional o regional deben dar prioridad a las necesidades sanitarias y no deben perjudicar las cadenas de suministro mundiales, entre otras cosas evitando medidas comerciales disruptivas y caóticas, como los aranceles elevados sobre los productos farmacéuticos.
Si bien las inversiones específicas en la producción nacional, orientadas por las necesidades sanitarias, pueden contribuir a aumentar la seguridad del suministro de medicamentos esenciales, el plan del presidente Trump para impulsar la capacidad farmacéutica nacional se basa en gran medida en la amenaza de aplicar aranceles elevados a las importaciones de productos farmacéuticos. Los caóticos oaranceles de Trump ignoran políticas más eficaces para aumentar la producción nacional y amenazan la capacidad mundial más amplia que puede apoyar el acceso oportuno a medicamentos asequibles para todos.
Mientras tanto, la administración Trump puede presionar a otros países para que compren productos estadounidenses como parte de acuerdos bilaterales de ayuda sanitaria, lo que podría obstaculizar el desarrollo de la capacidad de fabricación local o regional en los países en desarrollo y fijar precios inasequibles para los medicamentos.
4. Garantizar la seguridad, la eficacia y la calidad de los medicamentos.
Para ayudar a reducir la carga regulatoria y facilitar el acceso oportuno a los medicamentos, muchas autoridades reguladoras participan en iniciativas para armonizar los requisitos regulatorios y se basan en las evaluaciones realizadas por otros reguladores. Esto es sensato cuando los organismos públicos mantienen la flexibilidad necesaria para equilibrar la seguridad, la eficacia y el acceso en interés del. público. Sin embargo, los acuerdos comerciales pueden obligar a los países a aplicar determinadas normas de evaluación o a aceptar íntegramente las decisiones normativas externas, lo que puede limitar la capacidad de los organismos para actuar en interés público o adaptarse a las necesidades locales y puede crear vías para que las empresas dominantes influyan en los procesos de evaluación normativa de manera que se inhiba la discrecionalidad de cada parte en la administración de las políticas de regulación de los medicamentos.
Los países deben conservar su soberanía en la toma de decisiones sobre la regulación de los medicamentos.
Para cumplir con la obligación de certificar que los medicamentos son seguros, eficaces y de buena calidad, las autoridades reguladoras nacionales deben conservar su autonomía, al tiempo que adoptan prácticas —incluida la colaboración externa— que eviten duplicaciones innecesarias, faciliten el acceso oportuno y contribuyan al fortalecimiento de la regulación.
Los acuerdos comerciales no deben interferir en el espacio para regular en interés de la salud pública. El Acuerdo de Comercio Recíproco entre Estados Unidos y Malasia, negociado en respuesta a las amenazas arancelarias de Trump, exige que Malasia acepte la autorización previa de comercialización emitida por la FDA de Estados Unidos como prueba suficiente de que un producto farmacéutico fabricado en Estados Unidos cumple los requisitos de Malasia para la autorización de comercialización. Esta obligación suscita preocupación por la libertad de Malasia para determinar la mejor manera de abordar las decisiones reglamentarias —incluido cuándo y cómo recurrir a organismos reguladores externos— a fin de garantizar la seguridad y la eficacia de los productos. Además, la dependencia de organismos reguladores externos sin una colaboración y un intercambio de información, complementarios puede socavar el desarrollo de las capacidades reglamentarias de los países en desarrollo, lo que, a su vez, puede obstaculizar los esfuerzos por fortalecer la producción local y regional, que depende de organismos reguladores competentes.
5. Determinar libremente qué tratados internacionales son beneficiosos.
Los acuerdos comerciales bilaterales suelen exigir o instar a la adopción o el cumplimiento de tratados internacionales sobre propiedad intelectual. Muchos de estos tratados abordan los procedimientos que deben seguir los solicitantes de patentes para simplificar los procesos y requisitos en todos los países. La adhesión a los tratados no tiene por qué ser beneficiosa para los países en desarrollo, en particular las obligaciones que imponen nuevas cargas que exceden la capacidad local o entran en conflicto con otros intereses nacionales.
Los gobiernos deben resistirse a la coacción económica para adoptar tratados internacionales. Los gobiernos deben considerar si les conviene adoptar acuerdos internacionales, ya que estos pueden imponer requisitos adicionales potencialmente onerosos que van más allá de los exigidos por un acuerdo bilateral.
Los acuerdos comerciales recientes firmados bajo la administración Trump exigen que el país socio se adhiera y aplique plenamente muchos acuerdos adicionales. Por ejemplo, el Acuerdo de Comercio Recíproco entre Estados Unidos y Malasia y el Acuerdo de Comercio Recíproco entre Estados Unidos y Camboya mencionan 13 acuerdos internacionales sobre propiedad intelectual, incluidos los que afectan a la regulación de las patentes farmacéuticas.
6. Adherirse a procesos comerciales transparentes y responsables.
La administración Trump ha aprovechado el caos arancelario para obligar a otros países a participar en negociaciones comerciales secretas. Si bien la sociedad civil lleva mucho tiempo criticando la falta de transparencia de las negociaciones de los acuerdos de libre comercio, las negociaciones comerciales de la actual administración estadounidense han alcanzado un nuevo nivel de secretismo. Esta falta de rendición de cuentas aumenta el riesgo de que las empresas se apropien del proceso, lo que lleva a los gobiernos a comprometerse con disposiciones perjudiciales que podrían poner en peligro la salud. Las decisiones que afectarán a la salud de las personas no pueden negociarse a puerta cerrada ni estar dominadas por los intereses corporativos.
La política comercial y las negociaciones comerciales deben ser transparentes, participativas y responsables ante el público para garantizar que cualquier acuerdo refleje la opinión democrática y promueva el interés público.
Antes de que los gobiernos compartan los textos de sus propuestas en las negociaciones,
dichos textos deben publicarse en un proceso de comentario público oficial, y todos los
textos consolidados tras cada ronda de negociaciones también deben hacerse públicos para que los ciudadanos y los expertos de la sociedad civil puedan influir en su contenido antes de que se finalice el texto renegociado.
Los modelos favorables a las empresas y al monopolio, reforzados por el orden comercial actual y explotados por los poderosos, están fallando al mundo. La salud debe ser una garantía, no una moneda de cambio.
Organizaciones firmantes:
Public Citizen, Health GAP, ABAC/ONG Burkina Faso, Acción Internacional para la Salud Perú, ACHA-AFRICA, Advancing Synergy, Æqua, Africa Freedom of Information Centre, Alliance Sud, AMWU, Asia Pacific Research Network (APRN), Asociación Santa Micaela, Association for Proper Internet Governance, Association of Democratic Doctors (Verein demokratischer Ärzt*innen), Association of Women of Southern Europe, Australian Fair Trade and Investment Network, AVAC, Bekwarra hepatitis B support & advocacy initiative, BFLA, Both ENDS, Brazilian Interdisciplinary AIDS Association, BUKO Pharma-Kampagne Germany, Canadian Centre for Policy Alternatives, Cancer Alliance South Africa, CARAM Asia, Center for Economic and Policy Research, Centre for Health and Development Initiative Africa (CHDIA), Centre for Human Rights and Rehabilitation (CHRR), Coalition des organismes communautaires quebecois de lutte contre le sida (COCQ-SIDA), COALITION OF WOMEN LIVING WITH HIV AND AIDS, Communications Workers of America (CWA), Dr Uzo Adirieje Foundation (DUZAFOUND), European Trade Justice Coalition, Florida Physicians for Social Responsibility, Friends of Vulnerable Village Children Uganda – FVVC-Ug, Fundacion Huesped, Fundación IFARMA, GeneEthics, Georgetown University Center for Global Health Policy & Politics, Girl Rescue Foundation (GRF), Global Exchange, Global Health Council, Global humanitarian Progress GHP Corp, Global Justice Now (UK),
Good Health Community Programmes,
Health Action International,
Human Rights Defenders Coalition (HRDC),
ICHANGE,
Indonesia for Global Justice (IGJ)Institute for Policy Studies – Global Economy Project,
IRESCO,
JARRIDD International,
Just Futures Collaborative,
Just Treatment,
Kamukunji Paralegal Trust (KAPLET),
Kimirina,
LHL International Tuberculosis Foundation,
Mainline,
MARSAL,
Medical IMPACT,
Medicinas para la Gente Capitulo Latinoamerica,
Missionary Oblates of Mary Immaculate,
National Forum of People Living with HIV and AIDS Networks Uganda (NAFOPHANU),
Naturefriends Greece,
NETWORK Lobby for Catholic Social Justice,
Nigerian Women Agro Allied Farmers Association,
Open Markets Institute,
Partners In Health,
People’s Health Movement – North America,
Pharmaceutical Accountability Foundation,
PowerShift e.V,
PrEP4All,
Prescrire,
Public Eye,
Public Health Association of Australia,
Public Services International (PSI),
Resilient40,
Salud por Derecho,
Salud y Farmacos,
Sinatsisa Lubombo Women and girls Empowerment organization,
Social Security Works,
Southern and Eastern Africa Trade Information and Negotiations Institute (SEATINI),
Spark Street Advisors,
St. Hemmingways CBO,
T1International,
The Society for Children Orphaned By AIDS Inc. (SOCOBA),
The United Methodist Church – General Board of Church and Society,
Third World Network,
Trade Justice Education Fund,
Trade Justice Movement,
Trade Justice Network Canada,
Trade Justice New York Metro,
Traditional, Complementary and Integrative Healthcare Coalition,
Transform Trade,
Treatment Action Group,
Union Congolaise des Organisations des PvVIH,
Universities Allied for Essential Medicines,
Vikas Adhyayan KendraVoluntary Health Association of India,
La lectura de este artículo genera una sensación de indignación, cuando observamos la utilización del informe Especial 301, habitualmente utilizado como amenaza y sanción por EEUU para proteger sus intereses comerciales, para imponer a Argentina unas condiciones draconianas en la aplicación de los derechos de propiedad intelectual, las exclusividades de datos, o sobre la prórroga del periodo de protección de la patentes.
Pero la impunidad con la que somete EEUU a Argentina llega al extremo de obligar a la creación de un a fiscalía federal para proteger la propiedad intelectual. Sin duda, estos acuerdos aceptados por el gobierno de Milei convierten a Argentina en un estado vasallo.
Cuando Trump anunció sus infames «aranceles recíprocos» a principios de abril de 2025, hizo alarde de un informe que, según él, contenía «barreras no arancelarias» de otros países contra Estados Unidos. Esas «barreras no arancelarias» eran, en realidad, leyes y políticas que desagradan a las corporaciones estadounidenses, incluso aquellas que benefician el interés público, como las leyes que promueven medicamentos asequibles. Cuando ese mismo mes se publicó la «lista de vigilancia» anual más detallada sobre las leyes de propiedad intelectual, el Informe Especial 301, Public Citizen alertó de que esta lista anual de políticas de otros países, dirigida a las grandes farmacéuticas, podría servir como guía para las concesiones de las grandes farmacéuticas en las secretas negociaciones de Trump sobre «aranceles recíprocos».
El Acuerdo sobre Comercio e Inversión Recíprocos, firmado recientemente entre Estados Unidos y Argentina, lamentablemente, hizo precisamente eso. El acuerdo entre Estados Unidos y Argentina obligó a que Argentina hiciera concesiones explícitamente enumeradas en el Informe Especial 301, lo que representa un nuevo ejemplo en el uso de la política comercial estadounidense para presionar a los países a adoptar reglas de propiedad intelectual que expanden el poder monopólico de las grandes farmacéuticas a expensas del acceso a medicamentos asequibles.
Aumento de las quejas especiales 301 de Big Pharma
Si bien las normas comerciales acordadas internacionalmente en la Organización Mundial del Comercio (OMC) privilegian indebidamente los derechos de propiedad intelectual, dichos acuerdos al menos reconocen la necesidad de equilibrar los derechos de propiedad intelectual con los esfuerzos para garantizar un acceso asequible a los medicamentos y apoyar el interés público.
Sin embargo, el Informe Especial 301 tiene un largo historial de menoscabar estas consideraciones de interés público en favor de disposiciones maximalistas sobre propiedad intelectual que amplían el poder monopolístico. Expertos han cuestionado la legitimidad del proceso del Informe Especial 301, sugiriendo que la práctica del Informe Especial 301 de no justificar sus críticas y amenazar con sanciones unilaterales por prácticas que cumplen con el Acuerdo sobre los Aspectos de los Derechos de Propiedad Intelectual relacionados con el Comercio (ADPIC) de la Organización Mundial del Comercio viola las normas de justicia administrativa y las normas internacionales.
Ahora, con el acuerdo entre Estados Unidos y Argentina, la administración Trump ha llevado esta intimidación un paso más allá mediante medidas vinculantes basadas en cuestionables denuncias del Informe Especial 301, lo que amplía aún más la influencia inapropiada de las corporaciones sobre la política comercial estadounidense.
Esta es la primera vez que un acuerdo comercial estadounidense menciona explícitamente el Informe Especial 301 y exige a un país abordar una serie de denuncias de las grandes farmacéuticas planteadas en él.
El nuevo acuerdo exige a Argentina «adoptar medidas con prontitud para resolver plenamente los problemas identificados con respecto a Argentina en el Informe Especial 301 más reciente». El acuerdo incluye diversas concesiones relacionadas con la propiedad intelectual, entre ellas:
Directrices de patentes – Argentina está obligada a tomar medidas para derogar sus regulaciones nacionales relacionadas con las normas de patentes (Resoluciones Conjuntas No. 118/2012, No. 546/2012, No. 107/2012, y No. 283/2015).
Las resoluciones argentinas de 2012 y 2015 orientan la evaluación de las solicitudes de invenciones farmacéuticas y biotecnológicas. Estas directrices no modifican ni limitan los criterios de patentabilidad, sino que buscan garantizar la correcta aplicación de dichos estándares. Al derogar estas directrices, las empresas farmacéuticas tienen mayor probabilidad de obtener patentes basadas en productos o procesos excesivamente amplios o no innovadores, lo que extenderá aún más sus monopolios y restringirá el acceso a medicamentos asequibles.
Exclusividad de datos: Argentina debe elaborar un informe que analice la viabilidad, el alcance y los requisitos institucionales para implementar un régimen de protección de datos que sea compatible con los artículos 20.45 y 20.48 del Tratado entre Estados Unidos, México y Canadá (T-MEC). Todos los países parte del Acuerdo sobre los ADPIC deben proteger los datos no divulgados del uso comercial desleal. Argentina ofrece dicha protección. La búsqueda de exclusividad de datos por parte de Estados Unidos va más allá de la protección de datos, proporcionando un nivel adicional de monopolio, independiente de las patentes. El T-MEC exige cinco años de exclusividad de datos, durante los cuales los fabricantes de genéricos y las autoridades reguladoras no pueden utilizar los datos de una empresa original para otorgar la aprobación de comercialización de un medicamento genérico.
Prórrogas de patentes: Argentina debe elaborar un informe que analice las causas de los retrasos en el proceso de concesión de patentes, identifique las atribuibles a factores administrativos y evalúe la viabilidad legal de las prórrogas de la duración de las patentes en caso de retrasos irrazonables, y tomar medidas para reducirsignificativamente la tramitación de patentes, incluyendo las de invenciones biotecnológicas y farmacéuticas.
Las prórrogas de la duración de las patentes otorgan a las empresas años adicionales de monopolio, además del plazo estándar de 20 años. Si bien las extensiones de la vigencia de las patentes se asignan aparentemente para evitar retrasos, la variación en los períodos de revisión es normal en cada sistema y no indica una protección insuficiente de la propiedad intelectual. Extender la vigencia de las patentes retrasa el acceso a medicamentos genéricos y mantiene los precios altos.
Cumplimiento: Argentina debe establecer un organismo de coordinación para el cumplimiento de la propiedad intelectual y explorar la posibilidad de contar con una fiscalía federal especializada en propiedad intelectual.
Las empresas gozan de amplios recursos para hacer cumplir su propiedad intelectual. Las medidas de cumplimiento expansivas inclinan aún más la balanza de poder a favor de los intereses de la industria, por ejemplo, al ofrecer recursos más generosos por infracción y ampliar sus posibilidades de influencia. Este tipo de medidas corre el riesgo de frenar las acciones políticas legítimas destinadas a facilitar el acceso a los medicamentos.
Una plantilla preocupante
El acuerdo entre EE. UU. y Argentina revela la mentira tras la afirmación de Trump de que se enfrenta a las grandes farmacéuticas y reduce los precios de los medicamentos. Mientras distrae al público estadounidense de las acciones reales para bajar los precios con su vanidosa página web TrumpRx, que en realidad amenaza con aumentar los costos de los medicamentos para algunos usuarios en lugar de reducirlos, está utilizando negociaciones comerciales secretas para hacer el trabajo sucio de las grandes farmacéuticas y expandir su poder monopolístico a nivel mundial.
La Coalición para la Promoción de los Derechos de los Pacientes para el Acceso a los Medicamentos está llevando a cabo una revisión judicial de la Ley de Patentes de Indonesia de 2024, pidiendo el retorno de las protecciones cruciales contra el reverdecimiento de patentes
Este artículo es un buen ejemplo de la utilización de los procedimientos utilizados por la BigPharma de reverdecimiento (evergreening) de patentes para extender él monopolio y mantener él alto precios de los medicamentos.
La Coalición Indonesia para la Defensa de los Derechos del Paciente y el Acceso a Medicamentos está impulsando una revisión judicial de la Ley de Patentes renovada, promulgada en 2024, advirtiendo que socava el acceso a medicamentos esenciales y amenaza la sostenibilidad del programa del Seguro Nacional de Salud (JKN). Según la coalición, la nueva ley contiene disposiciones que amenazan el acceso público a medicamentos esenciales, en particular debido a la eliminación del Artículo 4(f), que anteriormente servía como protección contra el evergreenIng de patentes, una estrategia utilizada por las compañías farmacéuticas para extender los monopolios de patentes sobre medicamentos esenciales.
Activistas de la coalición declararon a People’s Health Dispatch que la disposición en cuestión había servido, durante muchos años, como una salvaguardia legal crucial y la base para que la sociedad civil se opusiera a las patentes, incluso en campañas relacionadas con los medicamentos bedaquilina y lenacapavir. Originalmente, el Artículo 4(f) estipulaba que los nuevos usos de productos conocidos, así como las nuevas formas de productos conocidos que sean químicamente similares y no proporcionen una mejora significativa en la eficacia, no podían protegerse como nuevas invenciones. El contenido principal del artículo «se asemejaba mucho al Artículo 3(d) de la Ley de Patentes de la India», señalaron los activistas.
«La eliminación de este artículo abre la puerta a peligrosos monopolios de patentes que perjudican a los pacientes», declaró Tony Richard Samosir, presidente de la Comunidad de Pacientes de Diálisis de Indonesia, en un comunicado que exponía las razones de la solicitud de revisión judicial. Estas preocupaciones están lejos de ser abstractas, dado que la perpetuación de patentes es una práctica perjudicial y bien documentada.
“Cuando se extienden las patentes sin una innovación clara, los precios de los medicamentos se mantienen altos y suponen una enorme carga para los pacientes, sus familias y el programa JKN”, añadió Arni Rismayanti, de la Fundación Indonesia para la Hipertensión Pulmonar. Se centró en el sildenafilo como un ejemplo concreto de cómo las compañías farmacéuticas manipulan las normas de patentes para aumentar sus ganancias a costa de la salud de los pacientes. Utilizado para tratar tanto la hipertensión pulmonar como la disfunción eréctil, el sildenafilo ahora puede patentarse en Indonesia bajo dos indicaciones independientes, “lo que permite que persistan los monopolios y que los precios se mantengan altos”, declaró Rismayanti.
Otro ejemplo planteado por activistas es la bedaquilina, un medicamento utilizado para tratar la tuberculosis multirresistente. «La patente principal de la bedaquilina en Indonesia expiró en 2023, pero cinco patentes secundarias han extendido el monopolio hasta 2036», señaló Irwandy Wijaya, de la Coalición Indonesia contra el SIDA. «Situaciones similares se han producido con medicamentos para el VIH/SIDA, la hepatitis C, el cáncer, la diabetes mellitus e incluso la COVID-19».
«Sin protecciones como el Artículo 4(f), estas estrategias pueden bloquear la producción de medicamentos genéricos y limitar gravemente el acceso público», advirtió Rahmat Maulana Sidik, director ejecutivo de Indonesia for Global Justice. «Con la eliminación del Artículo 4(f) de la actual Ley de Patentes, la supervisión pública sobre las patentes farmacéuticas que no cumplen con los estándares de patentes se ha visto significativamente debilitada», añadió.
Como parte del proceso judicial, la Coalición exige la restitución del Artículo 4(f) para proteger el derecho a la salud en Indonesia y resistir las presiones de las compañías farmacéuticas sobre el sector salud.
Uso de cookies
Este sitio web utiliza cookies para que usted tenga la mejor experiencia de usuario. Más info