
Revista nº 34 Noviembre – Diciembre 2024
Féix Bermejo Pareja 1 – María Victoria Zunzunegui 2
- Profesor emérito CIBERNED y honorífico UCM.
- Epidemióloga, viki.zunzunegui@gmail.com
Los autores no tienen nada que declarar.
Resumen
Introducción
La aprobación de lecanemab en EEUU como terapia de la enfermedad de Alzheimer (EA) y su no aprobación por la EMA (European Medical Agency) ha suscitado discusión. Presentamos un análisis crítico de esta terapia y una breve revisión nosológica de la EA.
Material y Métodos
Revisión de autores, con búsqueda de la literatura en bases biomédicas, fundamentalmente, en MEDLINE y Google Académico.
Resultados
Se efectúa una sucinta revisión de las terapias farmacológicas de la EA, y se analiza con más detalle la que considera que la fisiopatología de EA (hipótesis amiloide) está determinada por un depósito cerebral excesivo de β-amiloide, que requiere su eliminación mediante inmunización activa (vacunas) o pasiva (anticuerpos humanizados anti-β-amiloide). Estas estrategias no han tenido éxito (no evitan su progresión) y producen mínima (o dudosa) mejoría clínica no exenta de efectos adversos. Se analiza cómo el lecanemab, exponente de estos anticuerpos, no ha sido aprobado en Europa. Se cuestiona esta nosología sobre la EA por su fracaso terapéutico y se discute la asociación EA-envejecimiento y la reciente disminución de riesgo de demencia (y EA) en países ricos lo que apoya su prevención mediante intervenciones para reducir los factores de riesgo en cada etapa de la trayectoria vital.
Conclusiones
La no aprobación del lecanemab por la EMA se juzga positiva. Se requieren más estudios sobre las asociaciones: EA-envejecimiento y EA de múltiple patología. La disminución del riesgo de demencia en países ricos apoya la prioridad de su prevención frente a tratamientos farmacológicos de dudosa eficacia y seguridad.
Texto
1.) Introducción
Antes de comentar la terapia de la enfermedad de Alzheimer (EA) conviene recordar algunos puntos conceptuales e históricos de esta enfermedad.
La (EA) nació como premio del entonces más prestigioso psiquiatra del mundo, Emil Kraepelin, a uno de sus más brillantes discípulos, Alois Alzheimer, con una nueva enfermedad que llevaría este epónimo, EA, y con él la introdujo en su texto de psiquiatría de 1910 (Kraepelin, 1910) con muy pocos casos descritos. La nueva enfermedad era una demencia presenil con cambios histológicos peculiares, los ovillos neurofibrilares o degeneración neurofibrilar (DNF, acrónimo inglés), descritos por Alois Alzheimer en 1906; anteriormente esta demencia se asociaba con las placas seniles cerebrales (Alzheimer, 2011; Assal, 2019). Véase las figuras 1 y 2 en las que se representa las dos lesiones más características de esta enfermedad, aunque otras importantes son la pérdida neuronal y sináptica (Jellinger et al, 2020).
Figura 1. Ovillos neurofibrilares (NFT) en la enfermedad de Alzheimer
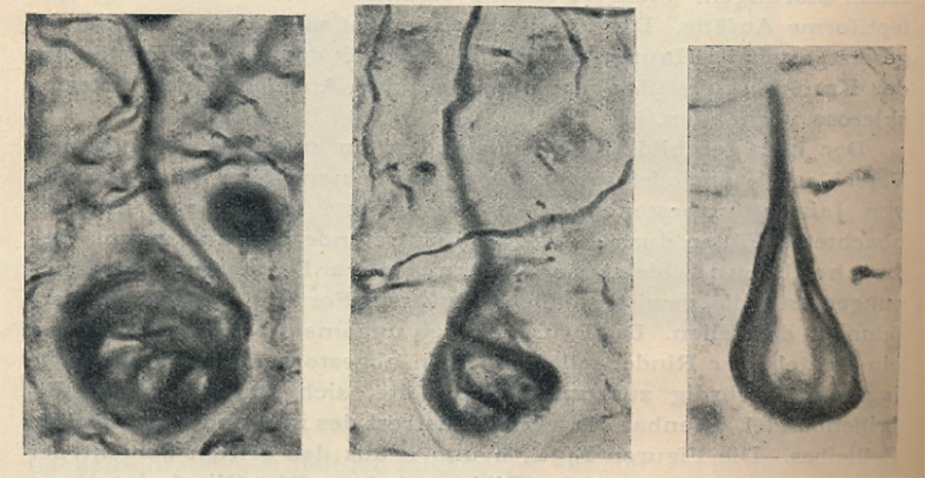
Imágenes de tres neuronas con ovillos neurofibrilares (o degeneración neurofibrilar –NDF) en su interior (ovillos de hilos gruesos en negro en el soma neuronal).
Foto reproducida del libro de Kraepelin de 1910 (véase Kraepelin, en la bibliografía)
Figura 2. Placas seniles neuríticas en la enfermedad de Alzheimer
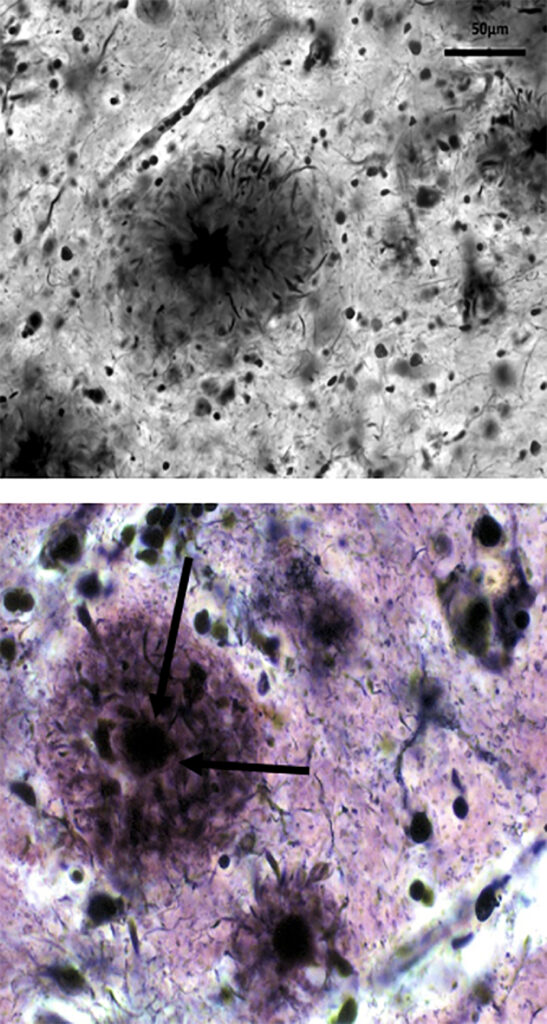
Imágenes de las placas seniles neuríticas. En la imagen superior se observa la placa senil (imagen redondeada en negro en el centro de la imagen) y se ven las neuritas alteradas que la rodean. Y en la foto inferior se visualiza muy bien en núcleo central de la placa (depósito de beta-amiloide), como indican las dos flechas, rodeada de neuritas.
Las imágenes pertenecen al archivo Cajal y han sido donadas por el Dr. Martínez, ex director del Instituto Cajal, para su reproducción no comercial.
(Las exponemos porque la calidad de las imágenes es superior a la de otros muchos neuropatólogos coetáneos).
Los coetáneos a Alois Alzheimer consideraron, la EA, una forma muy infrecuente de demencia presenil, pero ésta generó desde su nacimiento una indudable controversia de gran interés histórico y científico (Amaducci, 1996; Bermejo-Pareja y del Ser, 2024), que está fuera de la intención de este trabajo.
Berchold y Cotman (1998), examinando la historia de la demencia, comentan que desde la descripción de la EA hasta 1960 la atención médica a esta enfermedad fue casi puramente clínico-patológica, siendo el interés primordial discutir si la EA era o no una forma diferente de demencia senil.
Tras la segunda guerra mundial, el panorama de la demencia cambió considerablemente, sobre todo en EEUU: el envejecimiento poblacional, el incremento de su longevidad y su nuevo enriquecimiento en la ya afluente sociedad americana, determinó una influencia cada vez mayor de este grupo etario en todos los órdenes sociales. Según el historiador de la ciencia, Ballenger (2006), la sociedad americana aceptó la propuesta de diversas instancias médicas de la medicalización del declive cognitivo y de la demencia en la ancianidad, que había sido considerada “natural” durante siglos, convirtiéndola en una enfermedad. Tres brillantes médicos e intelectuales fueron los artífices de este cambio, Robert Butler, psiquiatra, premio Pulitzer, luchador contra el “ageism” (o edadismo, en su traducción en español) (Butler, 1969), y primer director del National Institute on Aging (NIA) norteamericano (Achenbaum, 2014). Otro gigante al que corresponde un gran mérito fue Robert Katzman, neurólogo e ideólogo de “changing view” (Katzman y Bick, 2000), que reconvirtió una rara demencia presenil en la demencia presenil y senil, cuya suma la hizo más frecuente, tanto que se convirtió en una causa mayor de mortalidad de los mayores en EEUU (Katzman, 1976; Kawas, 2009). A ellos se sumó un prestigioso neuropatólogo, Robert Terry, que le dio carta de naturaleza patológica a lo largo de décadas, aunque él consideraba que la causa verdadera de EA era un déficit sináptico (Terry RD et al, 2009 y Terry NA et al, 2019). En ese contexto histórico, el NIA consideró el tratamiento y curación de esta enfermedad como uno de los primus movens de su quehacer. El contexto científico europeo y mundial fue aceptando esta proposición poco a poco como demuestran varios estudios bibliométricos (Serrano-Pozo et al, 2017; Robert et al, 2020). Muy pocos médicos criticaron, entonces, la restringida postura del NIA, dejando en un lugar secundario la investigación del envejecimiento (Adelman, 1998).
Con el empuje teórico y económico del NIA y con la intervención entusiasta de la industria farmacéutica se han generado desde entonces múltiples intentos terapéuticos.
2.) Terapia farmacológica en el EA. Perspectiva histórica, y el caso del lecanemab
El NIA, siguiendo su política en investigación y “cura” de la EA le ha dedicado ingentes cantidades de dinero procedentes de este Instituto Federal americano (Fox, 1989), y posteriormente también en EEU, la Asociación de Alzheimer (última denominación de este lobby de apoyo a los enfermos de EA), y en general, en todo el mundo. Y ha habido logros indudables, como ejemplo el hallazgo de la degeneración de las neuronas colinérgicas del tronco cerebral descubierto por Whitehouse et al (1981), que sustentó morfológicamente el déficit de inervación colinérgica de la corteza cerebral en la EA, que, asociado a la previa verificación de la importancia de esta inervación con la memoria (Drachman y Leavitt, 1974; Bartus et al, 1982), permitió el desarrollo de fármacos colinomiméticos (aumentan la estimulación colinérgica), fundamentalmente inhibidores de la colinesterasa (fisostigmina, donepezilo, rivastigmina y otros) que han entrado en el arsenal terapéutico con éxitos pírricos en los ensayos clínicos, pero muy limitados en la práctica médica, pues solo producen mejoría clínica en un porcentaje reducido de casos, cuyo beneficio no suele superar dos años, y no desdeñables efectos secundarios, y desde luego, no curan ni el deterioro cognitivo progresivo de la EA ni atenúan su mortalidad (Sharma, 2019; Bermejo-Pareja y del Ser, 2024). Hay visiones algo más optimistas desde la perspectiva fisiopatológica de estos fármacos (Hampel et al, 2018).
El ingente desarrollo de fármacos contra la demencia y la EA (en la tabla 1 se expone una mínima muestra) tiene una línea preeminente que proviene de la genética, cuyo brillante desembarco en el siglo XXI, ha descrito nuestro acervo genético (ADN) y entrevisto la posibilidad de actuar en el metabolismo molecular de nuestro organismo. En el ámbito de la EA existen raros casos genéticos de EA familiar (1-5%), pues la mayoría (95-99%) son esporádicos (Bermejo-Pareja y del Ser, 2024). Y sus tres formas genéticas familiares (de comienzo precoz) se deben a alteraciones de los genes de APP, presenilina 1 y 2, cuya consecuencia molecular, en este ámbito, es determinar un exceso de producción de β-amiloide (βA), proteína que se acumula en el “core” central de las placas seniles de la EA (figura 2), y aunque dicho así parece simple, su acción metabólica cerebral es muy compleja (Steiner et al, 1999; Lanoiselée et al 2017; Bertram & Tanzi, 2020;) y generó la hipótesis amiloide (cascada amiloide) de la EA (Hardy & Higgins,1992), que predica, esencialmente, que el acúmulo excesivo de βA es tóxico para el cerebro y determina una cascada amiloide de eventos metabólicos que generan pérdida neuronal y demencia. Sus autores la siguen defendiendo (Selkoe y Hardy, 2016). Sobre esta hipótesis se ha basado la categorización diagnóstica y la investigación farmacológica de la EA durante más de un cuarto de siglo. Se ha intentado con varias estrategias terapéuticas eliminar el depósito excesivo de βA cerebral (Bermejo-Pareja y del Ser, 2024). Para ello se han utilizado modelos de animales transgénicos (Wirak et al, 1991), insertando genes humanos de pacientes con EA que, aunque no reproducen fielmente la EA, han permitido emplear fármacos que eliminan el βA de estos modelos (traspasados a los humanos) mediante inmunogénesis activa (vacunas), y posteriormente, mediante anticuerpos humanizados anti-βA (A-AβA) (Geylis et al, 2006). El bapineuzumab es un ejemplo paradigmático de A-AβA (Doody et al,2014) ¿Cuál ha sido el resultado?: descorazonador. La vacuna contra la EA no entró en el mercado por sus efectos adversos (Orgogozo et al, 2003). Pero el relevante estudio patológico de Nicoll et al (2019), ha mostrado que la inmunización activa se asoció con la desaparición de βA en el cerebro de los pacientes tratados con la vacuna, pero también ha constatado, que esta desaparición no mejoró su demencia que continuó progresando (así como las lesiones tipo tau) y la mayoría murió con demencia avanzada (19 de 22 casos). ¿Y qué ha pasado con los numerosos A-AβA?, pues han tenido un resultado análogo, pese a ensayos clínicos muy extensos y bien ejecutados, tanto en la EA genética (Salloway et al, 2021) como en la EA esporádica. Los A-AβA han demostrado que no alteran el curso progresivo de la demencia ni la mortalidad que esta enfermedad determina, aunque en algunos estudios se han evidenciado mejorías estadísticas en algunos test psicométricos, pero no de clara significación clínica (Foroutan et al, 2019; Ackley, et al, 2021; Lozupone et al, 2024). Y hay revisiones sistemáticas con métodos bayesianos que demuestran que estos fármacos no se han separado de la hipótesis nula (Richard et al, 2021), vaya, que no han demostrado efectos terapéuticos claros en la EA.
¿Y cómo es que se continua con la experimentación de estos fármacos y cómo se sostiene la hipótesis amiloide de la EA?, pues probablemente como se han sostenido muchos paradigmas científicos establecidos durante décadas. Pese a la demostración de una realidad palpable de corrientes críticas poderosas (Joseph et el 2001; Whitehouse y George, 2008; Høilund-Carlsen et al, 2020; Herrup, 2021; Lozupone et al, 2024) los paradigmas científicos son difíciles de desbancar como ha mostrado Kuhn (1970). Y en este caso las críticas han sido significativas (Huang et al, 2019; Nicoll et al, 2019; Mullane y Williams, 2020, Ackley et al, 2021; Kurkinen et al, 2023),y algunas feroces como las del decano de la Universidad de Oxford, AD Smith (2022), que ha llegado a sostener que no era ético continuar con la terapia anti-amiloide en la EA, y que la postura de muchos laboratorios de no dar acceso los ensayos a los investigadores, tampoco lo es. Pero la hipótesis amiloide ha generado una poderosa industria no solo de neurociencia básica y farmacológica sino de pruebas diagnósticas alentadas por la industria farmacéutica y acompañada por una tecnología cara y productiva económicamente, de diversos marcadores de EA (con neuroimagen sofisticada, léase RM, SPECT y PET) y determinaciones en sangre y LCR de las proteínas que no se circunscriben a la investigación médica como sería razonable sino que se utilizan en unidades clínicas más o menos especializadas, y cuyo basamento patológico y valor predictivo es escaso o dudoso (Ritchie et al, 2014; Bermejo-Pareja y del Ser, 2024), pero que mantienen en pie la hipótesis amiloide de la EA con argumentos probablemente más de política médica (mejores diagnósticos para el paciente) y farmacológica empresarial (inversión realizada en este campo), que puramente científica o de valor terapéutico para el paciente con esta enfermedad.
El caso del lecanemab es solo un ejemplo más de otros A-AβA ya utilizados, y que no difiere esencialmente de ellos. Después de la publicación en el New England Journal of Medicine de un único ensayo clínico que demostraba su eficacia en retrasar el deterioro cognitivo en la muestra del estudio (Van Dyck et al, 2023), fue aprobado rápidamente por la FDA (Food and Drug Administration) en julio de 2023 (Hoy, 2023), pese a la polémica suscitada sobre éste y otros estos fármacos semejantes (aducanumab, donanemab) (Mullane y Williams, 2020; Herrup 2021; Pang et al, 2023; Høilund-Carlsen et al, 2024), y hay autores que han dado la bienvenida al fármaco (Schiller et al, 2024), pero otros consideran que es una mala noticia (Kepp et al, 2023; Kurkinen, 2023). Sin embargo, recientemente la EMA (European Medicine Agency) ha rechazado su uso en Europa, alegando que su efecto de retraso del declive cognitivo no contrabalancea los efectos adversos serios asociados a su uso, y por consiguiente que se requieren más ensayos y de más larga duración.
Esta decisión inclina la balanza a favor de los críticos de la terapia con los actuales A-AβA. Y hemos de señalar la enorme atención en la literatura científica que ha determinado este fármaco con más de 350 citas en MEDLINE y, ya, con una revisión sistemática (Chowdhury y Chowdhury, 2023). Nos parece relevante la reacción de varios neurólogos (Burke et al, 2023) en Neurology (revista de la Academia Americana deNeurología), antes su aprobación en EEUU, y es preguntarse: “¿no necesitamos datos suficientes para estar seguros de que no es menos efectivo, mucho más dañino y 100 veces más costoso que el donepezilo?”. Y sostienen que se requieren más estudios y a más largo plazo para su uso. Nuestra opinión es semejante sobre los recientes A-AβA y es que, si el donepezilo no ha cambiado el porvenir de los pacientes con EA, pagar un coste de 100 veces mayor por fármacos no lo van a cambiar no parece una inversión sensata, ni siquiera en sociedades afluentes. No existe un elixir mágico contra la EA, y es muy difícil que lo haya en el contexto actual de conocimientos (Larson, 2014; Khachaturian, 2017) y la terapia anti-amiloide actual no lo es. También, el precursor del lecanemab, el donanemab, ha sido rechazado por NICE, la agencia de medicamentos del Reino Unido (Mahase, 2024). Pero ya hay nuevas estrategias futuras en este campo (Bhadane et al, 2024) y en la terapia de la EA (Zhang et al, 2024).
Tabla 1. Breve resumen de terapias farmacológicas en demencia y EA ¶
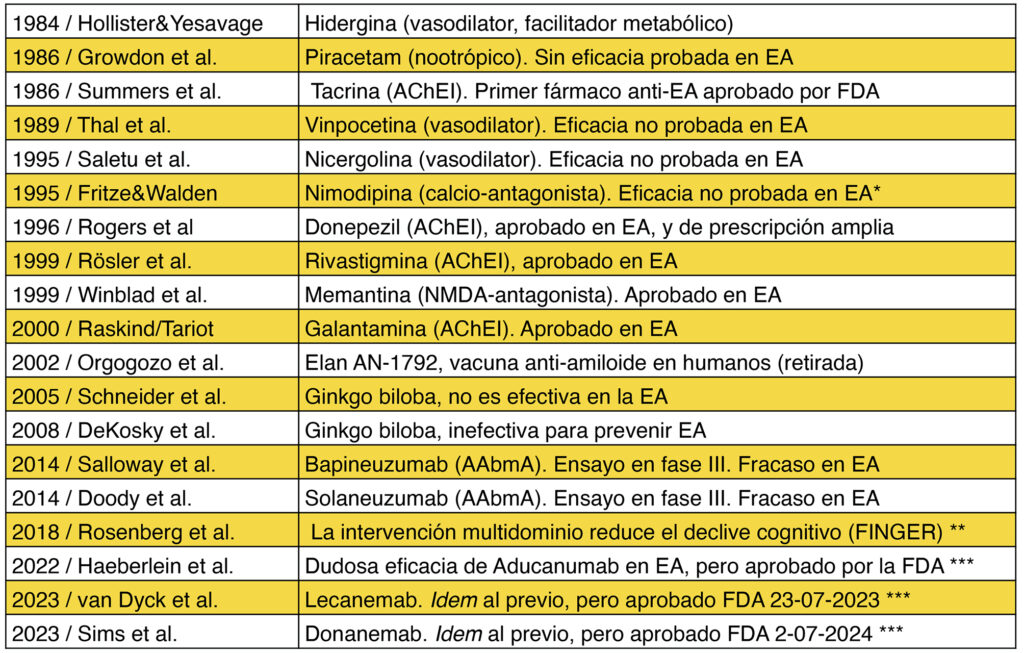
Abreviaturas en inglés:
AChEI, inhibidor de la acetilcolinesterasa; NMDA: N-metil-D-aspartato.;
AAbmA: anticuerpo monoclonal anti-amiloide-beta. FDA: Food & Drug Administration de EEUU
*Utilizada en demencia vascular y aprobada para el vasoespasmo por hemorragia subaracnoidea
** El estudio FINGER está realizado en pacientes sin demencia, pero con riesgo de padecerla
*** Estos fármacos reducen la carga de amiloide cerebral, pero pequeña mejoría en la EA según los autores de los estudios (tabla y texto) o muy dudosa para sus críticos (Høilund-Carlsen et al, 2024, y texto). La inmunoterapia activa no mejoró la demencia de la EA en 15 años (Nicoll et al, 2019).
Tomada y modificada de Bermejo-Pareja y del Ser, 2024, (trabajo en el que se citan las referencias de esta tabla y no se repiten en éste para no incrementar su listado).
3.). Demencia y envejecimiento
No es conveniente finalizar este trabajo sobre la EA y su terapia actual con resultados negativos por lo que conviene aclarar algunos puntos sobre este padecimiento. El primero sería sobre su génesis. Si la terapia basada en la hipótesis amiloide no ha conseguido fármacos curativos en más de 25 años ¿No hay otras hipótesis patogenéticas sobre la EA? Sí, hay más de 100, desde un criterio evolucionista (Rapoport, 1989), al infeccioso (Itzhaki et al, 2020), incluso alguno abstruso (Wostyn et al,2010), pero desde una perspectiva bibliométrica, la segunda hipótesis es el envejecimiento (Bermejo-Pareja y del Ser, 2024). Hipótesis que no es ni nueva ni definitiva. El envejecimiento es el más antiguo, estable y mayor factor de riesgo de la demencia, EA y de la mayoría de enfermedades neurodegenerativas (END), pero su relación desde una perspectiva fisiopatológica con estas enfermedades es desconocida (Yakner et al, 2008; Aron et al 2020). Tampoco son bien conocidas las causas del actual excepcional aumento de la supervivencia (¿mayor educación, hábitos saludables, nutrición, u otras?) en países desarrollados frente a los cazadores-recolectores actuales, que es superior a las 200 veces en ancianos (Vaupel, 2010; Burger et al, 2012).
La demencia en el anciano se achacó al envejecimiento, desde la civilización egipcia; y la medicina greco-romana se hizo eco de la misma, con alguna excepción como la de Cicerón (1924), con visión más benigna de la senectud, pues sugirió que una vida mental activa podría prevenir o posponer el declive cognitivo; visión que podemos firmar en la actualidad. Y hasta bien entrado el siglo XIX se hicieron muy pocos avances conceptuales en demencia porque ésta seguía considerándose una característica inevitable del envejecimiento (Berchold y Cotman, 1998). Y desde la perspectiva de la medicina naturalística, con Aristóteles a la cabeza, para la cual salud y enfermedad se distribuían de forma natural entre los seres vivos, es difícil de mantener que la demencia del anciano no sea sino una forma de envejecimiento exagerado del sistema nervioso. Sus egregios representantes actuales, ejemplo paradigmático, la “Bioestatistical theory” de Boorse y otros (Boorse, 2014; Kious, 2018), según la cual los fenómenos biológicos se distribuyen siguiendo parámetros de especie, edad y sexo con una distribución, digamos “gausiana”. Desde esa perspectiva, el declive cognitivo y la demencia del anciano sería una manifestación natural del envejecimiento, no una enfermedad (Brayne et al, 1995; Park et al, 2003; Singh-Manoux et al, 2010; Walhovd et al 2014) ¿Cómo considerar una enfermedad cuya incidencia crece de forma tan exponencial y análoga a la muerte en la naturaleza?, ¿Se puede considerar enfermedad una dolencia que afecta a un tercio de los mayores de 80 años, a un 25%-54% de los nonagenarios (Carrillo et al, 2008) y a quizá a más del 75% de los centenarios?, esto es la norma, no la excepción. (Brayne et al, 1995; Singh-Manoux et al, 2010; Breitner, 2014). Es cierto que las cifras de demencia y declive cognitivo aparente para los centenarios con buena salud son más benignas (45%), pero buena salud a esa edad no es la norma (Ailshire et al, 2024). Desde la perspectiva de la medicina naturalística no se podría sostener que la EA es una enfermedad.Pero estamos en tiempos y paradigmas de medicina normativa, y las normas médicas han establecido que sí lo es… Y, aun siguiendo esta ruta, muchos autores sostienen que declive cognitivo, demencia y EA están indisolublemente unidos al envejecimiento (Brayne, 1995, Lock, 2013; Ferrer, 2022), incluso a nivel básico, celular (Liu, 2022; Lau et al 2023). El estudio del envejecimiento y su relación con las enfermedades cerebrales como se lamentaba Adelman (1998), ha sido indebidamente postergado por la ciencia médica. Hay indicios de que en el futuro las cosas no van a ser así. Muchas entidades sociales están liderando el estudio del envejecimiento celular y molecular (Anti-Aging de Jeff Bezzos, Calico Lab, Hevolution Foundation, y otros) (Bermejo-Pareja y del Ser, 2024).
4. ¿Necesita la EA una redefinición?
Aunque esta consideración se aparta algo del tema central de este artículo, conviene exponer que el siglo XXI ha traído dos cambios conceptuales importantes en el ámbito de la demencia y EA.
1). Patología de la demencia y EA. Las series patológicas que actualmente recogen cerebros de los muy ancianos han mostrado, que su patología más frecuente son las causas mixtas: EA más lesiones vasculares, solas ambas, o asociadas a múltiples patologías tipo END (Schneider et al, 2007; Wharton et al 2023) entre las que destaca la LATE, acrónimo en inglés de “limbic-predominant age-related TDP-43 encephalopathy” (Nelson et al, 2019), establecida por innovaciones de las técnicas patológicas. La patología de la demencia del anciano (y EA) ha mostrado una gran multicausalidad patológica lo que dificulta que fármacos aislados tengan efectos terapéuticos en la demencia y EA del anciano (Jellinger, 2020; Nichols et al, 2023).
2). La disminución de la incidencia de demencia en países ricos occidentales (disminución no verificada en orientales como Japón o Corea del Sur) es una noticia muy positiva; el riesgo de demencia en las dos-tres últimas décadas ha disminuido debido (quizás) al efecto de la mayor educación de las poblaciones, mejora de las condiciones de vida desde la infancia, y terapia de los trastornos crónicos (Satizabal et al, 2016; Wu et al, 2017).
Estos dos hechos han propiciado: a) que la EA sea considerada más como un síndrome multicausal que como una enfermedad bien definida (Richard y Brayne, 2010). Zaven Khachaturian (2015), exdirector de estudios extramurales del NIA, y de la principal revista del campo de las demencias, lo sustenta; b) la prevención de la demencia del anciano se ha mostrado como una posibilidad plausible. Así, una comisión de la prestigiosa revista, “Lancet “, en varias publicaciones ha identificado 14 factores de riesgo, fundamentalmente clínicos, a los que se puede atribuir el 45% de los casos de demencia (Livingstone et al, 2017, 2020 y 2024). Otros estudios que analizan aspectos más amplios que los puramente clínicos, inciden en aspectos del desarrollo (maternos e infantiles), económicos y sociales a lo largo de la vida; y elevan esa cifra: 66% de contribución de factores ambientales en el desarrollo de demencia y EA (Xu et al, 2015; Bermejo-Pareja,2018; Weiss et al, 2020).
El estudio FINGER ensayo clínico multidimensional (Ngandu et al, 2015; Rosenberg et al, 2017) ha suscitado esperanzas porque los sujetos incluidos en la intervención preventiva retrasaban su declive cognitivo versus controles. En España, en Beasain (Euskadi) se ha realizado una experiencia piloto FINGER con resultados prometedores (Tainta et al, 2024). Estos estudios invitan a creer en la efectividad de intervenciones poblacionales para disminuir el riesgo de demencia como ha mostrado una revisión sistemática (Castro et al, 2023). Estos hechos animan a la implementación de políticas de salud pública locales y estatales y de los servicios de salud de cada comunidad (Bermejo-Pareja et al, 2016).
Lo que parece claro es que en el ámbito de la demencia del anciano se requiere de un nuevo paradigma científico en el que la investigación sobre el envejecimiento y la educación (reserva cognitiva) debieran ocupar un lugar central, así como las estrategias de prevención individual y poblacional. Es de desear que la investigación genética, epigenética y farmacológica faciliten el camino a un envejecimiento más satisfactorio con menor declive cognitivo, demencia y EA (Hachinski, 2023; Bermejo-Pareja y del Ser, 2024; Zhang et al, 2024).
5.). Conclusiones
La breve revisión de la terapia farmacológica de la EA pone de manifiesto la importancia de la hipótesis amiloide en el desarrollo de fármacos que eliminen el βA de del cerebro mediante inmunogénesis activa (vacunas), y posteriormente, mediante anticuerpos humanizados anti-βA. Ambas estrategias han fracasado tanto en la EA genética como en la esporádica. El lecanemab es un fármaco que no difiere de otros A-AβA ya utilizados, pero que ha suscitado gran controversia al ser aprobado por la FDA americana y no por la EMA. Juzgamos positiva la actitud de la EMA desde la perspectiva del paciente, de la ciencia y de la sociedad (coste elevado).
La discusión sobre el constructo nosológico actual de la EA concluye afirmando que la EA debe considerarse más como un síndrome multicausal muy ligado al envejecimiento que como una enfermedad sensu estricto.
Los datos recientes que indican una disminución del riesgo de demencia del anciano (y EA) apoyan la necesidad de una estrategia de prevención durante toda la trayectoria vital mediante una acción holística, médica y social y opuesta a la aprobación y acceso universal a tratamientos farmacológicos de dudosa eficacia y seguridad.
Referencias bibliográficas
Achenbaum WA. Robert N. Butler, MD (January 21, 1927-July 4, 2010): visionary leader. Gerontologist. 2014; 54:6-12. doi: 10.1093/geront/gnt015.
Ackley SF, Zimmerman SC, Brenowitz WD, et al. Effect of reductions in amyloid levels on cognitive change in randomized trials: instrumental variable meta-analysis. BMJ. 2021; 372:n156. doi: 10.1136/bmj.n156.
Adelman RC. The alzheimerization of aging: A brief update Exp Gerontol. 1998; 33: 155–157. doi: 10.1016/s0531-5565(97)00057-0.
Ailshire JA, Beltrán-Sánchez H, Crimmins EM. Becoming centenarians: disease and functioning trajectories of older US Adults as they survive to 100. J Gerontol A Biol Sci Med Sci. 2015; 70:193-201. doi: 10.1093/gerona/glu124.
Alzheimer A. Über eigenartige Krankheitsfälle des späteren Alters. Z Ges Neurol Psychiatr. 1911; 4:356-385. (Versión en inglés de Förstl H, Levy R. On certain peculiar diseases of old age. Hist Psychiatry. 1991; 2:71-101. doi: 10.1177/0957154X9100200505).
AmaducciL.Alzheimer’soriginalpatient. Science 1996, 274,328.https://doi.org/10.1126/science.274.5286.328a.
Aron L, Zullo J, Yankner BA. The adaptive aging brain. Curr Opin Neurobiol. 2022; 72:91-100. doi: 10.1016/j.conb.2021.09.009.
Assal F. History of dementia. Front Neurol Neurosci. 2019; 44:118-126. doi: 10.1159/000494959.
Bhadane P, Roul K, Belemkar S, Kumar D. Immunotherapeutic approaches for Alzheimer’s disease: Exploring active and passive vaccine progress. Brain Res. 2024;1840:149018. doi: 10.1016/j.brainres.2024.149018.
BallengerJF.; Self, Senility, and Alzheimer’s Disease in Modern America: A History;TheJohnHopkinsUniversityPress:Baltimore,MD,USA,2006.
Bartus RT, Dean RL III, Beer B and Lippa AS: The cholinergic hypothesis of geriatric memory dysfunction. Science. 1982; 217: 408‑417. doi: 10.1126/science.7046051.
BerchtoldN,CotmanC.EvolutionintheconceptualizationofdementiaandAlzheimer’sDisease:Greco-Romanperiodtothe1960s. Neurobiol. Aging 1998, 19,173–189.https://doi.org/10.1016/s0197-4580(98)00052-9.
Bermejo‐Pareja F. Alzheimer: Prevention from Childhood. Lamberth Academic Publishing; Mauritius. 2018.
Bermejo-Pareja F, Llamas-Velasco S, Villarejo-Galende A. Alzheimer’s disease prevention: A way forward. Rev Clin Esp (Barc). 2016; 216:495-503. (English & Spanish). doi: 10.1016/j.rce.2016.05.010.
Bermejo-Pareja F, Del Ser T. Controversial past, splendid present, unpredictable future: A brief review of Alzheimer disease history. J Clin Med. 2024; 13:536. doi: 10.3390/jcm13020536.
Bertram L, Tanzi RE. Genomic mechanisms in Alzheimer’s disease. Brain Pathol. 2020; 30:966-977. doi: 10.1111/bpa.12882.
Boorse C. A second rebuttal on health. J Med Philos. 2014; 39: 683–724.doi:10.1093/jmp/jhu035.
Brayne C, Gill C, Paykel ES, Huppert F, O’Connor DW. Cognitive decline in an elderly population–a two wave study of change. Psychol Med. 1995; 25:673-83. PMID: 7480446.
Breitner JC. What should we do if we were wrong and Alzheimer was right? Int Psychogeriatr. 2014; 26:3-6. doi: 10.1017/S1041610213001749.
Burger O, Baudisch A, Vaupel JW. Human mortality improvement in evolutionary context. Proc Natl Acad Sci U S A. 2012; 109:18210-4. doi: 10.1073/pnas.1215627109.
Burke JF, Kerber KA, Langa KM, Albin RL, Kotagal V. Lecanemab: looking before we leap. Neurology. 2023; 101:661-665.doi:10.1212/WNL.0000000000207505
Butler RN. Age-ism: another form of bigotry. Gerontologist 1969; 9:243-6. doi: 10.1093/geront/9.4_part_1.243.
Carrillo-Alcalá ME, Bermejo-Pareja F. [Dementia in nonagenarians. Systematic review of population-based studies with Spanish data]. Rev Neurol 2008; 47:347-54 PMID: 18841545.
Castro CB, Costa LM, Dias CB, et al. Multi-Domain Interventions for Dementia Prevention – A Systematic Review. J Nutr Health Aging. 2023; 27:1271-1280. doi: 10.1007/s12603-023-2046-2.
CicerónMT. De senectute. Tricastela. Madrid. 2001 (Versión en latín y español).
Chowdhury S, Chowdhury NS. Novel anti-amyloid-beta (Aβ) monoclonal antibody lecanemab for Alzheimer’s disease: A systematic review. Int J Immunopathol Pharmacol. 2023; 37:3946320231209839. doi: 10.1177/03946320231209839.
Doody RS, Farlow M, Aisen PS; Alzheimer’s Disease Cooperative Study Data Analysis and Publication Committee. Phase 3 trials of solanezumab and bapineuzumab for Alzheimer’s disease. N Engl J Med. 2014; 370:1460. doi: 10.1056/NEJMc1402193.
Drachman DA, Leavitt J. Human memory and the cholinergic system. A relationship to aging? Arch Neurol. 1974; 30:113-21. doi: 10.1001/archneur.1974.00490320001001.
European Medicine Agency (EMA). Refusal of the marketing authorization for Leqembi (lecanemab). https://www.ema.europa.eu/en/medicines/human/EPAR/leqembi (Accedido el 28 de octubre de 2024).
Ferrer I. Alzheimer’s disease is an inherent, natural part of human brain aging: an integrated perspective. Free Neuropathol. 2022; 3:3-17. doi: 10.17879/freeneuropathology-2022-3806.
Fox P. From senility to Alzheimer’s disease: the rise of the Alzheimer’s disease movement. Milbank Q. 1989; 67:58-102. PMID: 2682166.
Geylis V, Steinitz M. Immunotherapy of Alzheimer’s disease (AD): from murine models to anti-amyloid beta (Abeta) human monoclonal antibodies. Autoimmun Rev. 2006; 5:33-9. doi: 10.1016/j.autrev.2005.06.007.
Hachinski V; Dementia Prevention/Brain Health Initiative. We are preventing some dementias now-But how? The Potamkin lecture. Alzheimers Dement. 2023; 19:1067-1072. doi: 10.1002/alz.12770.
Hampel H, Mesulam MM, Cuello AC, et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain. 2018; 141:1917-1933. doi: 10.1093/brain/awy132.
Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science 1992; 256: 184-5. doi: 10.1126/science.1566067.
Herrup K. How not to Study a Disease. The Story of Alzheimer’s disease. The MIT Press. Cambridge. Mass. 2021.
Høilund-Carlsen PF, Barrio JR, Werner TJ, Newberg A, Alavi A. Amyloid hypothesis: The Emperor’s new clothes? J Alzheimers Dis. 2020; 78:1363-1366. doi: 10.3233/JAD-200990.
Høilund-Carlsen PF, Alavi A, Barrio JR, et al. Donanemab, another anti-Alzheimer’s drug with risk and uncertain benefit. Ageing Res Rev. 2024; 99:102348. doi: 10.1016/j.arr.2024.102348.
Hoy SM. Lecanemab: First Approval. Drugs. 2023; 83:359-365. doi: 10.1007/s40265-023-01851-2.
Huang YM, Shen J, Zhao HL. Major clinical trials failed the amyloid hypothesis of Alzheimer’s disease. J Am Geriatr Soc. 2019; 67:841-844. doi: 10.1111/jgs.15830.
Itzhaki RF, Golde TE, Heneka MT, Readhead B. Do infections have a role in the pathogenesis of Alzheimer disease? Nat Rev Neurol. 2020; 16:193-197. doi: 10.1038/s41582-020-0323-9
Jellinger KA. Neuropathology of the Alzheimer’s continuum: an update. Free Neuropathol. 2020 Nov 11;1:1-32. doi: 10.17879/freeneuropathology-2020-3050.
Joseph J, Shukitt-Hale B, Denisova NA, Martin A, Perry G, Smith MA. Copernicus revisited: amyloid beta in Alzheimer’s disease. Neurobiol Aging. 2001; 22:131-46. doi: 10.1016/s0197-4580(00)00211-6
Katzman R. Editorial: The prevalence and malignancy of Alzheimer disease. A major killer. Arch Neurol. 1976; 33:217-8. doi: 10.1001/archneur.1976.00500040001001
Katzman R, Bick K. Alzheimer Disease. The Changing View. Academic Press. San Diego. 2000.
Kawas CH. Robert Katzman, MD. J Alzheimers Dis. 2009; 17:1-3. doi: 10.3233/JAD-2009-1039.
Kepp KP, Sensi SL, Johnsen KB, et al. The anti-amyloid monoclonal antibody Lecanemab: 16 cautionary notes. J Alzheimers Dis. 2023; 94):497-507. doi: 10.3233/JAD-230099.
KhachaturianZS.Prospectsforeffectivetreatmentofthedementia-alzheimersyndrome:ArenewedOdysseyinsearchofthemagicelixir. J Prev Alzheimers Dis. 2017; 4:215–217. doi:10.14283/jpad.2017.42.
Khachaturian ZS, Khachaturian AS. Politics of science: progress toward prevention of the dementia–Alzheimer’s syndrome. Mol Aspect Med. 2015; 43‐44:3‐15. doi: 10.1016/j.mam.2015.06.001.
Kious BM.Boorse’stheory of disease: (why) do values matter? J Med Philos. 2018; 43: 421-438. doi:10.1093/jmp/jhy012
Kraepelin E. Psychiatrie: Ein Lehrbuch Fuer Studierende Und Aerzte. Verlag von Johann Ambrosius Barth. Lepzig.1910. Descrición en inglés de Schorer CE. Historical essay: Kraepelin’s description of Alzheimer’s disease. Int J Aging Hum Dev. 1985; 21:235-8. doi: 10.2190/gnq1-gdux-eptl-0f2l (el libro original en alemán se encuentra en la biblioteca del I. Cajal en Madrid).
Kuhn TS. The Structure of Scientific Revolutions. Second Edition. The University of Chicago. 1970.
Kurkinen M. Lecanemab (Leqembi) is not the right drug for patients with Alzheimer’s disease. Adv Clin Exp Med. 2023; 32:943-947. doi: 10.17219/acem/171379.
Kurkinen M, Fułek M, Fułek K, Beszłej JA, Kurpas D, Leszek J. The amyloid cascade hypothesis in Alzheimer’s disease: Should we change our thinking? Biomolecules. 2023; 13:453. doi: 10.3390/biom13030453.
Lanoiselée HM, Nicolas G, Wallon D, et al. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLoS Med. 2017; 14:e1002270. doi: 10.1371/journal.pmed.1002270.
Larson EB. Prevention of late-life dementia: No magic bullet. Ann Intern Med. 2018; 168:77-79. doi: 10.7326/M17-3026.
Lau V, Ramer L, Tremblay MÈ. An aging, pathology burden, and glial senescence build-up hypothesis for late onset Alzheimer’s disease. Nat Commun. 2023; 14:1670. doi: 10.1038/s41467-023-37304-3.
Livingston G, Sommerlad A, Orgeta V, et al Dementia prevention, intervention, and care. Lancet. 2017; 390:2673-2734. doi: 10.1016/S0140-6736(17)31363-6.
Livingston G, Huntley J, Sommerlad A, et al. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet 2020; 396:413-446. doi: 10.1016/S0140-6736(20)30367-6.
Livingston G, Huntley J, Liu KY, et al. Dementia prevention, intervention, and care: 2024 report of the Lancet standing Commission. Lancet. 2024; 404:572-628. doi: 10.1016/S0140-6736(24)01296-0.
LiuRM.Aging,cellularsenescence,andAlzheimer’sdisease. Int J Mol Sci. 2022; 23:1989.https://doi.org/10.3390/ijms23041989.
Lock, M. The Alzheimer Conundrum: Entanglements of Dementia and Aging; Princeton University Press: Princeton, NJ, USA, 2013.
Lozupone M, Dibello V, Sardone R et al. Lessons learned from the failure of solanezumab as a prospective treatment strategy for Alzheimer’s disease. Expert Opin Drug Discov. 2024; 19:639-647. doi: 10.1080/17460441.2024.2348142.
Mahase E. NICE rejects Alzheimer’s drug donanemab owing to cost and «significant health risks». BMJ. 2024; 387:q2342. doi: 10.1136/bmj.q2342.
Mullane K, Williams M. Alzheimer’s disease beyond amyloid: Can the repetitive failures of amyloid-targeted therapeutics inform future approaches to dementia drug discovery? Biochem Pharmacol. 2020; 177:113945. doi: 10.1016/j.bcp.2020.113945.
Nelson PT, Dickson DW, Trojanowski JQ, et al. Limbic-predominant age-related TDP-43
encephalopathy (LATE): consensus working group report. Brain 2019: 0; 1–25. doi: 10.1093/brain/awz099.
Ngandu T, Lehtisalo J, Solomon A, et al.. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): a randomized controlled trial. Lancet. 2015; 385: 2255–63.
Nicoll JAR, Buckland GR, Harrison CH, et al. Persistent neuropathological effects 14 years following amyloid-β immunization in Alzheimer’s disease. Brain. 2019; 142:2113-2126. doi: 10.1093/brain/awz142.
Orgogozo JM, Gilman S, Dartigues JF, et al. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology 2003; 61:46-54. doi: 10.1212/01.wnl.0000073623.84147.a8.
Pang M, Zhu L, Gabelle A, et al. Effect of reduction in brain amyloid levels on change in cognitive and functional decline in randomized clinical trials: An instrumental variable meta-analysis. Alzheimers Dement. 2023; 19:1292-1299. doi: 10.1002/alz.12768.
Park HL, O’Connell JE, Thomson RG. A systematic review of cognitive decline in the general elderly population. Int J Geriatr Psychiatry. 2003; 18:1121-34. doi: 10.1002/gps.1023.
Rapoport SI. Hypothesis: Alzheimer’s disease is a phylogenetic disease. Med Hypotheses 1989; 29:147-50. doi: 10.1016/0306-9877(89)90185-0
Richard E, den Brok MGHE, van Gool WA. Bayes analysis supports null hypothesis of anti-amyloid beta therapy in Alzheimer’s disease. Alzheimers Dement. 2021; 17:1051-1055. doi: 10.1002/alz.12379
Richards M, Brayne C. What do we mean by Alzheimer’s disease? BMJ 2010; 341:c4670. doi: 10.1136/bmj.c4670.
RitchieC.SmailagicN,Noel-StorrAH et al. PlasmaandcerebrospinalfluidamyloidbetaforthediagnosisofAlzheimer’sdiseasedementiaandotherdementiasinpeoplewithmildcognitiveimpairment(MCI). Cochrane Database Syst. Rev. 2014,CD008782.doi:10.1002/14651858.cd008782.pub4.
Robert C, Wilson CS, Lipton RB, Arreto CD. Evolution of the research literature and the scientific community of Alzheimer’s disease from 1983-2017: A 35-year survey. J Alzheimers Dis. 2020; 75:1105-1134. doi: 10.3233/JAD-191281.
RosenbergA,NganduT,RusanenM, et al.Multidomainlifestyleinterventionbenefitsalargeelderlypopulationatriskforcognitivedeclineanddementiaregardlessofbaselinecharacteristics:TheFINGERtrial. Alzheimer’s Dement. 2017;14:263–270.https://doi.org/10.1016/j.jalz.2017.09.006.
Satizabal CL, Beiser AS, Chouraki V, Chêne G, Dufouil C, Seshadri S. Incidence of dementia over three decades in the Framingham Heart Study. N Engl J Med. 2016; 374:523-32. doi: 10.1056/NEJMoa1504327.
Schiller ER, Silverglate BD, Grossberg GT. Profiling lecanemab as a treatment option for Alzheimer’s disease. Expert Rev Neurother. 2024; 24:433-441. doi: 10.1080/14737175.2024.2333970..
Schneider JA, Arvanitakis Z, Bang W, Bennett DA. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 2007; 69:2197-204. doi: 10.1212/01.wnl.0000271090.28148.24.
Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. 2016; 8:595-608. doi: 10.15252/emmm.201606210.
Sharma K. Cholinesterase inhibitors as Alzheimer’s therapeutics (Review). Mol Med Rep. 2019; 20:1479-1487. doi: 10.3892/mmr.2019.10374.
Singh-Manoux A, Kivimäki M. The importance of cognitive aging for understanding dementia. Age (Dordr). 2010; 32:509-12. doi: 10.1007/s11357-010-9147-7.
Smith AD. Anti-amyloid trials raise scientific and ethical questions. BMJ. 2021; 372:n805. doi: 10.1136/bmj.n805.
Steiner H, Capell A, Leimer U, Haass C. Genes and mechanisms involved in beta-amyloid generation and Alzheimer’s disease. Eur Arch Psychiatry Clin Neurosci. 1999; 249:266-70. doi: 10.1007/s004060050098.
Tainta M, Ecay-Torres M, de Arriba M, et al. GOIZ ZAINDU study: a FINGER-like multidomain lifestyle intervention feasibility randomized trial to prevent dementia in Southern Europe. Alzheimers Res Ther. 2024; 16:44. doi: 10.1186/s13195-024-01393-z.
Terry RD, Masliah E, Salmon DP, et al. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991; 30:572-80. doi: 10.1002/ana.410300410.
Terry N, Masliah A, Overk C, Masliah E. Remembering Robert D. Terry at a time of change in the world of Alzheimer’s disease. J Alzheimers Dis. 2019; 70:621-628. doi: 10.3233/JAD-190518.
Vaupel JW. Biodemography of human ageing. Nature. 2010; 464:536-42. doi: 10.1038/nature08984.
Van Dyck CH, Swanson CJ, Bateman RJ, et al. Lecanemab in Early Alzheimer’s Disease. N Engl J Med 2023; 388:9-21. doi:: 10.1056/NEJMoa2212948.
Weiss J, Puterman E, Prather AA, WareEB, Rehkopf DH. A data-driven prospective study of dementia among older adults in the United States. PLoS ONE. 2020 15: e0239994. doi : g/10.1371/journal.pone.0239994.
Wharton SB, Simpson JE, Ince PG, et al. Insights into the pathological basis of dementia from population-based neuropathology studies. Neuropathol Appl Neurobiol. 2023; 49:e12923. doi:10.1111/nan.12923.
Walhovd KB, Fjell AM, Espeseth T. Cognitive decline and brain pathology in aging–need for a dimensional, lifespan and systems vulnerability view. Scand J Psychol. 2014; 55:244-54. doi: 10.1111/sjop.12120.
Whitehouse PJ, George D. The Myth of Alzheimer’ Disease. What you aren’t being told about today’s most dreaded diagnosis. St Martin’s Griffin. New York. 2008.
Whitehouse PJ, Price DL, Clark AW, Coyle JT, DeLong MR. Alzheimer disease: evidence for selective loss of cholinergic neurons in the nucleus basalis. Ann Neurol. 1981; 10:122-6.
Wirak DO, Bayney R, Ramabhadran TV, et al. Deposits of amyloid beta protein in the central nervous system of transgenic mice. Science. 1991; 253:323-5. doi: 10.1126/science.1857970.
Wostyn P, Audenaert K, De Deyn PP. Alzheimer’s disease: cerebral glaucoma? Med Hypotheses 2010; 74:973-7. doi: 10.1016/j.mehy.2009.12.019.
Xu W, Tan L, Wang HF, et al. Meta-analysis of modifiable risk factors for Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2015; 86:1299-306. doi: 10.1136/jnnp-2015-310548.
Wu YT, Beiser AS, Breteler MMB, et al. The changing prevalence and incidence of dementia over time – current evidence. Nat Rev Neurol. 2017; 16:327-339. doi: 10.1038/nrneurol.2017.63.
Yankner BA, Lu T, Loerch P. The aging brain. Annu Rev Pathol. 2008; 3:41-66. doi: 10.1146/annurev.pathmechdis.2.010506.092044.
Zhang J, Zhang Y, Wang J, Xia Y, Zhang J, Chen L. Recent advances in Alzheimer’s disease: Mechanisms, clinical trials and new drug development strategies. Signal Transduct Target Ther. 2024; 9:211. doi: 10.1038/s41392-024-01911-3.