- Joan Ramon Laporte, Mercedes Zurita, AVITE, Oriol Güell y NoGracias, reciben los Premios de la Asociación por un Acceso Justo al Medicamento. Manuel Rico (Investigate Europe), reconocimiento especial.
- Palabras de Soledad Cabezón, presidenta de la AAJM: ”La actual política farmacéutica mata; atenta contra el Derecho Humano a la salud”.
Revista nº 34 Noviembre – Diciembre 2024
Comisión de Redacción de la rAJM

El catedrático de Farmacología Joan Ramon Laporte, la Dra. Mercedes Zurita (Hospital Universitario Puerta de Hierro), la Asociación de Víctimas de la Talidomida en España (AVITE), el periodista Oriol Güell (El País), la Organización Civil Internacional NoGracias y el periodista Manuel Rico, de la coalición europea Investigate Europe, recibieron el pasado 23 de noviembre los Premios de la Asociación por un Acceso Justo al Medicamento (AAJM).
El acto, que se celebró en el Ayuntamiento de Noblejas (Toledo), presidido por su alcalde, Agustín Jiménez Crespo, y la presidenta de la AAJM, Soledad Cabezón. Durante el mismo, se expresaron numerosas muestras de apoyo a las víctimas de la DANA, tanto de la comunidad autónoma de Valencia como de Castilla-La Mancha. Intervinieron también el Juan José Rodríguez Sendin, médico de Noblejas y ex presidente de la AAJM. El presidente de la Organización Médica Colegial. Tomás Cobo, envió un video de aliento a la labor de la AAJM.
Agustín Jiménez expresó su agradecimiento a la AAJM por volver a Noblejas como escenario de estos. En su intervención, hablo de enfermedad y pobreza, dos situaciones -afirmó- cuya alianza no es una casualidad porque “la enfermedad se ceba en la falta de recursos, en el hambre y en las carencias de un sistema sanitario insuficiente en recursos humanos y terapéuticos cuya pérdida progresiva de capacidad de respuesta -afirmó- no debemos seguir tolerando”.
“Por eso -añadió- apoyamos a la Asociación por un Acceso Justo al Medicamento porque pensamos que solo los ciudadanos bien informados y motivados serán capaces de cambiar una situación que origina la mayor morbimortalidad en el mundo junto con el mal reparto de la riqueza y la pobreza”. Aludió al crecimiento “desmesurado y sin control” del precio de los medicamentos que “ha agravado esta situación sin que los gobiernos, no solo no hagan nada para evitarlo, sino que sean cómplices de la misma”.
Premiados
Los Premios AAJM 2024, que este año celebran su II edición coincidiendo con el séptimo aniversario de la asociación, reconocen la labor de profesionales, instituciones, organizaciones y medios de comunicación que han destacado por su labor en defensa de la asistencia sanitaria de cobertura universal, del acceso justo a medicamentos y vacunas para todo el mundo y en la denuncia del uso abusivo de las leyes de propiedad intelectual que impiden el acceso a los medicamentos necesarios.

Joan Ramon Laporte, premio “Personalidad destacada en el ámbito político / sanitario” y Soledad Cabezón, presidenta de la AAJM. Acceso a vídeo PINCHANDO AQUÍ..
Joan Ramon Laporte, catedrático emérito de Farmacología de la Universidad Autónoma de Barcelona, recibió el premio “Personalidad destacada en el ámbito político/ sanitario” por su labor de denuncia entono a los ensayos clínicos y su crítica permanente y fundada a las prácticas de las empresas farmacéuticas.
Joan Ramon Laporte, tras agradecer el premio, resaltó la labor de la AAJM, “clave en el debate permanente sobre las políticas farmacéuticas”, y en especial la Revista de la AAJM que “ha contribuido de manera decisiva a su visibilidad”.
En su intervención habló de los riesgos por las fallas en los procesos de regulación y autorización de los medicamentos y dijo que “la mayoría de los nuevos medicamentos de precios exorbitantes no tienen pruebas convincentes de eficacia, y mucho menos de seguridad”. Asimismo, hizo referencia al sistema de patentes que “ha favorecido -dijo- la innovación comercial, pero no ha dado lugar a grandes avances en terapéutica” y se mostró partidario crear compañías farmacéuticas públicas, capaces de dar viabilidad al conocimiento generado desde el propio sistema sanitario.

Dra. Mercedes Zurita, premio “Mejor labor de una Institución científico-sanitaria” y Ángel María Martín Fernández-Gallardo, vicepresidente de la AAJM. Acceso a vídeo PINCHANDO AQUÍ. .
Dra. Mercedes Zurita, investigadora y responsable de la Unidad de Terapia Celular del Hospital Universitario Puerta de Hierro, recibió el premio a la “Mejor labor de una Institución científica-sanitaria”, por su trabajo de investigación de la terapia NC1 para pacientes con lesión medular, primer tratamiento de terapia avanzada y fabricación no industrial aprobado en España por la AEMPS que se ha convertido en uno de los referentes en investigación clínica.
Para Mercedes Zurita, este premio supone, “tanto para mí como para el resto del equipo de investigación que tengo el honor de dirigir, un reconocimiento a nuestro trabajo y a la dedicación que hemos tenido durante tantos años”.
La Dra. Zurita calificó las Terapias Avanzadas como la “medicina del futuro” y, tras destacar el gran potencial científico de la investigación en hospitales, cuestionó el hecho de que “muchos de desarrollos científicos que se hacen nuestros hospitales caigan en manos de farmacéuticas que al final son las que se llevan el reconocimiento y el dinero que generan las investigaciones realizadas al menos en parte con dinero público”. “Luego toca -añadió- comprar esas patentes a un alto precio para ponerlas a disposición de los pacientes por lo que el estado paga dos veces por lo mismo”.

Juan José Rodríguez Sendín, expresidente de la AAJM y Andrés Vizcaino que recogió el premio “Mejor labor de una Asociación de Pacientes” en nombre de la Asociación de Víctimas de la Talidomina de España (AVITE). Acceso a vídeo PINCHANDO AQUÍ. .
La Asociación de Víctimas de la Talidomida en España (AVITE) fue reconocida en la categoría de “Mejor labor de una Asociación de Pacientes”, por su lucha contra el imperio farmacéutico Grünenthal para conseguir el reconocimiento de las consecuencias que sufrieron a raíz del consumo de la talidomida en embarazadas, lo que provoco en los años 50 y 60 toda una tragedia con miles de casos de malformaciones congénitas y miles de muertes en todo el mundo.
Para Andrés Vizcaino, este premio a AVITE supone el “reconocimiento a una labor humanitaria en favor de un colectivo con severas discapacidades, ocasionadas por los efectos adversos de la talidomida”. Afirmo que “la evidencia de lo que provocó este medicamento ha servido para modificar legislaciones sobre autorización y comercialización de nuevos medicamentos, en favor de la protección de los pacientes, y los potenciales efectos secundarios”.
En su lucha constante para ayudar a las víctimas, Andrés Vizcaino, dijo que AVITE trabaja para que las ayudas no sean gravadas con un IRPF medio del 47%, lo que deja estas ayudas en la mitad y para que se aplique la Sentencia del Tribunal Supremo que ha dado la razón a AVITE, y reconoce otros 150 nuevos afectados, y que estos perciban las ayudas.

Fernando Lamata, expresidente de la AAJM y Oriol Güel premio “Mejor labor de información y divulgación”. Acceso a vídeo PINCHANDO AQUÍ.
El periodista Oriol Güell, responsable de temas de sanidad de El País, fue premiado en la categoría “Mejor labor de información y divulgación” por su labor informativa, rigurosa e investigadora en torno a sanidad pública, a la salud, y a la investigación, producción, comercialización y riesgos de diferentes medicamentos.
Oriol Güell agradeció a la AAJM por premiar su trabajo informativo como el de escribir sobre medicamentos y la industria farmacéutica que, “en ocasiones -afirmó- no es sencillo dada la complejidad de algunos temas y los fuertes intereses de los agentes implicados”.
Destacó la tendencia de los poderes públicos hacia una mayor transparencia sobre los precios de los medicamentos, cuya clave es “la voluntad política de todos los países y organismos internacionales implicados”. También hizo referencia al sistema de patentes y dijo que “a menudo las patentes no incentivan la innovación y el incentivo pasa conseguir extensiones poco justificadas de las patentes, a lo que se suma que la investigación proviene de centros púbicos”, por lo que “hay muchas razones que apuntan a que el sistema de patentes requeriría una revisión”.

Roberto Sánchez, que recogió en nombre de NoGracias el premio “Mejor labor de una Organización” y Roberto Sabrido, expresidente de la AAJM. Acceso a vídeo PINCHANDO AQUÍ.
Organización Civil Internacional NoGracias, recibió el premio a la “Mejor Labor de una Organización”, en la persona de su presidente Roberto Sánchez, por su labor crítica y de denuncia constante, a lo largo de más de 15 años, en defensa de la asistencia sanitaria universal y la transparencia en torno al buen gobierno de los medicamentos, así como la labor divulgativa para que los médicos no se dejen influir por los laboratorios.
Roberto Sánchez agradeció el premio a NoGracias por lo que supone un reconocimiento al trabajo de organizaciones que “representamos al interés público y que confrontamos con el gigante médico-industrial, a pesar de contar con pocos medios, problemas en el acceso a información clave y numerosas dificultades en el ejercicio de nuestra actividad”.
El presidente de NoGracias hizo referencia a la falta de transparencia en los precios de los medicamentos y tras preguntarse “¿Cómo puede ser que con nuestros impuestos se estén financiando bajo secreto comercial carísimos medicamentos? y ¿En qué otro sector de políticas es esto posible?”, afirmó que “el secreto es un instrumento que impone la industria para seguir manteniendo esos precios en la seguridad de que el escrutinio público sería incapaz de soportar y mantenerlos”.

Agustín Jiménez Crespo, alcalde de Noblejas y Manuel Rico, “Reconocimiento especial”. Acceso a vídeo PINCHANDO AQUÍ.
El periodista Manuel Rico, miembro de la coalición europea de periodistas de investigación Investigate Europe recibió un reconocimiento especial por toda su labor de investigación sobre el poder de las farmacéuticas y, en especial, por su trabajo para esclarecer lo ocurrido durante la primera ola de la COVID-19 en las residencias y apoyar a las familias de las víctimas.
Manuel Rico agradeció el reconocimiento al “trabajo colectivo que hemos realizado en Investigate Europe” y a la asociación que otorga este premio, que “trabaja para lograr justicia en un ámbito tan esencial como el acceso a los medicamentos para conseguir una sociedad más justa, algo por lo que estudié Periodismo, porque quería aportar mi granito de arena en ese camino”.
También se pronunció sobre la necesidad de poner fin a los elevados precios de los medicamentos y los beneficios desorbitados de la industria farmacéutica y dijo que para ello es preciso que haya “información, transparencia y mucha pedagogía” porque “la gente desconoce la realidad del mundo farmacéutico”. “Tener la voluntad de superar el problema, lograr suficiente influencia allí donde se toman las decisiones y llegar a otros ámbitos de la sociedad civil para evidenciar que son prácticas inaceptables”, es el camino, según Manuel Rico, para poner fin a esta situación.

Soledad Cabezón, presidenta de la AAJM.
Soledad Cabezón, presidenta de la AAJM, agradeció al Ayuntamiento de Noblejas, en especial a su alcalde, su “compromiso social manifiesto con la atención sanitaria a los ciudadanos” y su apoyo a la AAJM en “esta justa reivindicación para garantizar el derecho al acceso a la salud mediante el acceso a los medicamentos”.
Tras felicitar a los premiados, Soledad Cabezón expuso las características que definen la política farmacéutica actual (una intervención que de la que por el interés de su contenido recogemos a continuación un amplio resumen).
La actual política farmacéutica mata; atenta contra el Derecho Humano a la salud.
Según la OMS, un tercio de la población mundial carece de la posibilidad de obtener medicamentos esenciales (casi 2.000 millones de personas; mueren 10 millones de personas cada año, mayoritariamente en las poblaciones más pobres, y afecta más a personas de color, mujeres y niños).
En España y en la UE, una media de un 4% de la población, según Eurostat, declara no poder acceder a los tratamientos prescritos, donde el copago es un factor clave.
Es tal la problemática del acceso a los medicamentos que es hoy uno de los grandes retos de salud mundial. La Agenda 2030 lo recoge en su ODS nº3.
Los avances científicos y tecnológicos solo alcanzan a un 14% de la población del mundo, a EEUU, Japón y Europa, que consumen más del 80% de los medicamentos, mientras que el 86% restante de la población mundial sólo puede permitirse acceder al 20% de los medicamentos que se fabrican.
La OMS señala que un 93 % de la mortalidad evitable por enfermedades sucede en los países en vías de desarrollo, pero solo un 5 % de la investigación se dirige a problemas de salud de estas áreas por falta interés económico de la industria o falta de recursos (las denominadas enfermedades olvidadas, como son la tuberculosis o la malaria, erradicadas).
En cuanto a las enfermedades consideradas no transmisibles, causante principal de la mortalidad mundial, como la diabetes y las enfermedades cardiovasculares, la OMS señala también que solo alrededor de una quinta parte de las personas con hipertensión en el mundo reciben un tratamiento eficaz.
Podemos decir que la pandemia del VIH continúa más de 30 años después de que impulsase la Declaración de DOHA. Y es que, aunque se haya tratado a más de 22 millones de personas portadoras de VIH en el mundo, cada año casi un millón de personas sigue muriendo a causa del VIH/sida por falta de acceso a los tratamientos de última generación, como al lenacapavir.
Más cercano nos suena el que la OMS cifre en más de 40 millones de muertes innecesarias, o exceso de mortalidad, por falta de acceso a la vacuna contra el Covid19 durante la pandemia.
Por otro lado, la resistencia antimicrobiana es responsable de más 35000 muertes al año en la UE por falta de I+D en nuevos antibióticos. En 2050, de seguir la tendencia, un aumento continuo de la resistencia antimicrobiana podría dar lugar a 10 millones de muertes al año en todo el mundo, con una reducción del 2 al 3,5% del PIB mundial embargo, pero es que desde la década de los 90 del siglo pasado no se ha descubierto una nueva familia de antibióticos.
En definitiva, la industria investiga atendiendo a las reglas del mercado, siendo su fin el de los beneficios económicos y no los sociales, binomio incompatible con el derecho al acceso a la salud.
La política farmacéutica arruina nuestro sistema sanitario; amenaza su salud.
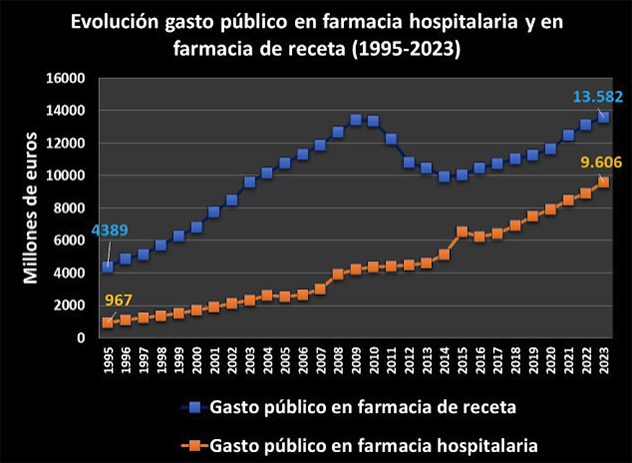
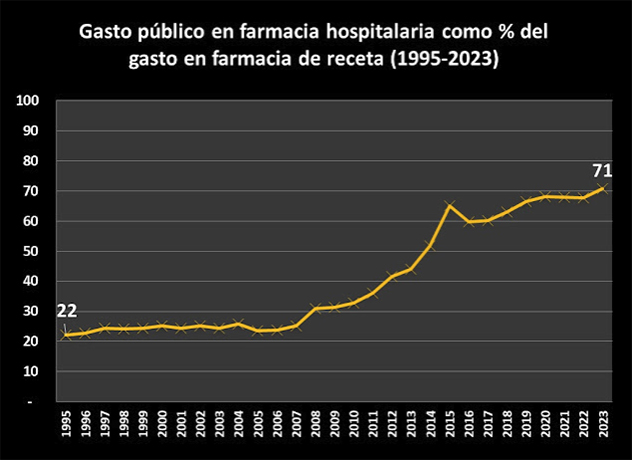
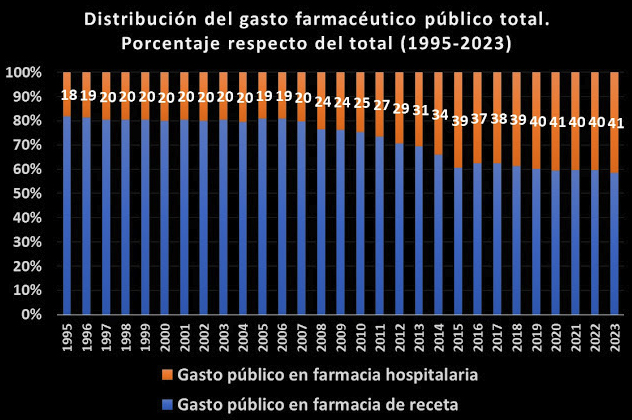
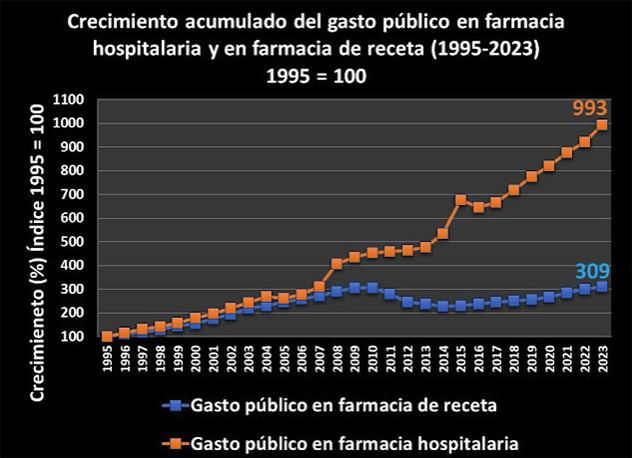
El negocio farmacéutico supone un 20% del gasto sanitario en la OCDE; según Lamata: «En España se gasta más en medicamentos que en todo la Atención Primaria. Entre noviembre de 2021 y 2022 aumentó un 5% el gasto farmacéutico. Eso son 616 millones de más en gasto farmaceútico ambulatorio que es lo que costaría contratar a 6.000 médicos «.
La OCDE ha proyectado que el crecimiento del gasto en salud de fuentes públicas será el doble del crecimiento promedio de los ingresos gubernamentales (2,6% y 1,3% respectivamente) entre 2019 y 2040. En consecuencia, el gasto en salud de fuentes públicas se prevé que alcance el 20,6% de los ingresos gubernamentales, un aumento de 4,7 puntos porcentuales con respecto a 2018.
Según IQVIA, a nivel mundial, en 2017 el gasto farmacéutico fue un 56% más que en 2007, con un crecimiento consecutivo del 12-14% cada quinquenio y se prevé que este será del 38% a 2028.
Este gasto se debe en gran parte a los altos precios de los nuevos medicamentos autorizados en las últimas décadas hasta llegar a ser inasequibles para muchos ciudadanos, incluso europeos, y amenazar seriamente la sostenibilidad de los sistemas sanitarios nacionales.
Los precios de los medicamentos no han dejado de incrementarse desde comienzos del siglo XX, pero especialmente a partir de finales de la segunda mitad con la aplicación de los derechos de propiedad intelectual creando monopolios de mercado, cuya generalización en el mundo se realiza a partir de 1995 con los Acuerdos de los ADIPC de la OMC.
Si el costo medio mensual del tratamiento contra el cáncer en 1960 era de 100 dólares en EEUU, pasó a más de 10.000 en 2012 y a más de 100.000 dólares por paciente y año para los nuevos tratamientos. Sin que, por otro lado, se haya acompañado de mejoras médicas proporcionales.
Precios, por otro lado, injustificados. Como recoge la publicación de Lamata y Gálvez, que indican que según las cifras publicadas por EFPIA (Federación Europea de Industria Farmacéuticas) sobre el gasto farmacéutico en la UE en 2016 de 179.535 millones de euros, si se hubiesen vendido a precio de genérico el coste habría sido un 41,5% de lo pagado, a lo que, si se le añadiese el gasto en I+D declarado por la misma fuente, EFPIA, de 26.913 millones, estiman que en la UE-28 hubo un exceso de gasto de 72.850 millones de euros sobre el precio de venta.
La política farmacéutica actual es sólo un modelo de negocio que adolece de ética.
Con, al menos, un 20% de retorno de inversión, el sector farmacéutico es uno de los sectores más lucrativos del mundo con el doble de margen beneficio en la UE que el resto de los sectores industriales.
Para muestra un botón; ya conocen ustedes como el recientemente comercializado inicialmente como tratamiento para la diabetes, pero utilizado masivamente para abordar la obesidad, ha colocado en riqueza a la empresa danesa Novonordisk por encima del PIB de su propio país Dinamarca en 2023. Pero en 2024, los ingresos netos suponen ya un 67% más que en 2023, con un 26% más de ganancias netas. Y lo que se espera…., a tenor de las noticias diarias sobre las múltiples posibles ampliaciones de indicaciones, incluido el alzheimer!, aunque adolezcan de los estudios adecuados, como sabe bien el catedrático en farmacología aquí presente, el Dr. Joan Ramón Laporte.
Mientras, el coste en general del desarrollo de estos y otros medicamentos se estiman en torno a 200 millones de dólares de media, según los datos facilitados por entidades sin ánimo de lucro, aunque la industria los cifra en 2870 millones de dólares, si bien se niegan a facilitarlos de forma transparente.
Datos más cuestionados aún para justificar los precios de los medicamentos si se tiene en cuenta la tendencia actual de adquisición por las industrias Big Pharma de lo que se ha venido a denominar empresas derivadas, de origen académico en su mayoría y financiación pública o por entidades sin ánimo de lucro, que, a la fecha, encabezan EEUU y el Reino Unido.
En el Reino Unido, la Universidad de Cambrigde ha desarrollado el 34% de ellas, mientras que la Universidad de Oxford el 13% de las empresas derivada, y cuyos costes de adquisición imputan como costes de investigación por parte de la industria.
Un ejemplo reciente y escandaloso lo tenemos en la vacuna contra el Covid de la empresa Moderna. El NIH (Instituto de Investigación Nacional de EEUU) acordó licenciar su tecnología a Moderna por 400 millones de dólares, lo que le revirtió a la empresa en 36 mil millones de dólares en ventas globales. Por su parte, el gobierno de EE.UU. invirtió al menos 31.900 millones de dólares para desarrollar, producir y comprar vacunas de ARNm contra el covid-19.
La política farmacéutica adoptada en el siglo XX es innecesaria para mejorar la salud.
Este modelo especulativo de la industria farmacéutica no fue siempre así, sino que es muy reciente, como el propio neoliberalismo.
El origen de la industrialización del producto farmacéutico se inicia a partir del siglo XIX, primero por parte de farmacéuticos y luego de personas procedentes de otra formación, como los químicos, que irán descubriendo nuevas sustancias que tendrán una aplicación terapéutica y a las que se les aplicarán procedimientos industriales con el desarrollo de la industria química; se produce así un tránsito progresivo de la fabricación artesanal de medicamentos en la rebotica de las farmacias a una elaboración industrial manufacturada del producto a escala. Durante el siglo XIX se van sucediendo descubrimientos de numeras sustancias químicas con aplicación terapéutica.
En América, un primer impulso de la industria farmacéutica se producirá tras la Guerra Civil (1861-1865), alcanzando un mayor grado de I+D, junto a Reino Unido, a raíz de la I Guerra Mundial (1914 -1918), debido a la interrupción del suministro de productos químicos alemanes y suizos. Aunque será la II Guerra Mundial (1939-1945) la que suponga el verdadero punto de inflexión hacia la industria farmacéutica moderna basada en I+D.
La II Guerra Mundial impulsa la fabricación a gran escala de la penicilina por las compañías farmacéuticas estadounidenses, cuya utilidad fue descubierta por el escocés Dr. Fleming en 1928, pero que hasta entonces se obtenía del cultivo de Penicillium en botes de cristal. El descubrimiento de la estreptomicina en el año 1944 por Marker supuso un enorme avance para el tratamiento de la tuberculosis con su desarrollo por el laboratorio Merck.
A este desarrollo industrial y surgimiento de numerosas moléculas se asiste sin la necesidad de derechos de propiedad industrial, aunque sí con un crucial apoyo público.
De nuevo, recordemos como en la historia reciente el apoyo público no ha sido menos crucial para impulsar la tecnología ARNm utilizada en las vacunas contra la Covid19, sobre la cual ya estaba realizada la investigación básica cuando irrumpe la pandemia; una I+D pública que servirá para el desarrollo de futuras y el impulso de nuevas terapias líneas de tratamiento. Pero también el desarrollo de su aplicación por parte del sector industrial contó con la inestimable financiación pública, cifrada en más de 5.800 millones de dólares, pero ahora ya bajo los derechos de propiedad intelectual, con miles de millones de beneficios para las empresas, pagados por los gobiernos (31.900 millones de dólares de factura en el caso de EEUU; aún estamos pendiente de los datos de la UE que la Comisión Europea se resiste a publicar) y que han dejado un exceso de mortalidad de 40 millones de personas en el mundo.
A finales de la primera mitad del siglo XX, en EEUU, la industria farmacéutica surgida en las reboticas, impulsada con el desarrollo de la industria química y posteriormente con el apoyo público durante las épocas de guerra, aunque no sólo, y que da lugar al surgimiento de empresas como Lilly o el auge de Novartis, comienza a presionar para que se apliquen los derechos de propiedad intelectual al medicamento.
A partir de finales de la década de los años sesenta del siglo XX, comienza a apreciarse una fuerte tendencia de los países desarrollados, en general, por incorporar los medicamentos en los sistemas de patentabilidad para proteger a sus industrias. En los países en vía de desarrollo será más tarde, a finales de la década de los 1980, con las negociaciones de la Ronda Uruguay.
En Europa, los derechos de propiedad intelectual se empiezan a incorporar al medicamento más tarde; en 1967 en Alemania, en 1968 en Francia, 1977 en Suiza y 1980 en Italia.
En España, la Ley de Propiedad Industrial de 1929 lo prohibía expresamente y no será hasta 1986, tras la firma del Tratado de Adhesión a la Comunidad Europea, cuando se introduzca la patente del medicamento en nuestro país.
Sin embargo, en 1987, la Secretaría de la OMPI preparó un informe sobre los sectores tecnológicos excluidos de la patentabilidad en las diferentes legislaciones nacionales, siendo habitual encontrar al farmacéutico entre los 19 sectores habitualmente excluidos. Este informe servirá de base para lo que vino después, los Acuerdos de los ADIPC, adoptados en 1995 en la OMC, en el contexto de la globalización económica, que vendrán a establecer a nivel internacional normas comunes en torno al comercio y extenderán y ampliarán la aplicación de forma obligatoria de los derechos de propiedad intelectual a todas las áreas del conocimiento, incluyendo, ahora sí, a los medicamentos.
La patente del producto farmacéutico supondrá un monopolio para la industria farmacéutica, con exclusividad de mercado durante años, eliminando la competencia, con el fin de recuperar la inversión económica realizada en el desarrollo del producto y estimular así la I+D. Queda claro que este sistema ha contribuido claramente en que el sector farmacéutico se haya convertido a día de hoy en el más competitivo de la UE, como también el que la factura farmacéutica ha ido creciendo hasta representar entorno al 20% del gasto en salud en los países de la UE y la OCDE y una media 1,5 % respecto del PIB.
Sin embargo, no parece que haya estimulado la I+D en cantidad ni en calidad. Un estudio llevado a cabo por la Universidad de Harvard en 2009 señalaba que el número de moléculas nuevas no conocidas no sólo era mayor previamente a los Acuerdos de los ADIPC, sino que tras su aprobación el número de nuevas sustancias fue menor e incluso, viendo la evolución de la serie, pareciera como si se hubiese desincentivado la calidad innovativa, no recuperándose nunca el número de nuevas sustancias de la década de los 70.
La OCDE también publica otra serie en la que la media de autorizaciones por la FDA de nuevos productos, concepto más amplio que el de nuevas sustancias, se ha mantenido estable desde 1980 a 2014, en torno a 100 al año.
En 2015, la consultora IQVIA estimaba en una media de 46 aprobaciones de nuevos medicamentos al año por la FDA en la última década y así lo preveía hasta 2028. Del mismo modo, el Institute for Healthcare Informatics (IMS) no observa variaciones significativas desde 2005 a 2018 en la aprobación de nuevas sustancias. En cuanto a las cifras en la UE publicadas, los nuevos medicamentos se aproximarían a los 40. Sin embargo, si se limitase a las sustancias realmente nuevas, sería la mitad.
Sin embargo, lo llamativo es como el número de nuevas sustancias dista mucho con el número de patentes de productos farmacéuticos concedidas en estos años por la UPSTO y la OEP, especialmente tras la entrada en vigor de los ADIPC.
Según los datos de la base de datos de la OCDE, la aprobación de los Acuerdos de los ADIPC supuso un llamativo incremento del número de aplicaciones de modificaciones o variaciones de productos previamente autorizados, en total se han registrado 77.645 aplicaciones de variaciones tipo I y 20.853 de tipo II, y de patentes en general, contabilizadas en miles desde 1995, y no de nuevas sustancias. Es decir, este acuerdo vino a dar lugar a un modelo de negocio muy lucrativo, pero nada innovador y, por lo tanto, innecesario e injustificado, a diferencia del mantra mantenido por los defensores del modelo vigente y la necesidad de los derechos de propiedad intelectual para estimular la I+D farmacéutica.
En definitiva, mientras que el número de patentes de medicamentos se cuentan en miles, se evidencia una falta de innovación genuina. Detrás de este alto número de patentes se encuentra la estrategia denominada “maraña de patentes” como modelo de negocio que mediante pequeñas modificaciones de los productos para alargar la vida de la patente se perpetúan las ganancias impidiendo la entrada de los genéricos al mercado. La Comisión Europea en su informe de 2008, único en el que estudió y publico él funcionamiento del mercado farmacéutico de forma global en la UE pero que sigue tan vigente como entonces, señaló que se daba el caso incluso de que un medicamento obtuviese 1300 patentes y cifraba que más del 78% de las patentes no se trataban de nuevos medicamentos, infravalorado este porcentaje si se atendiese al número de nuevas moléculas o principios activos.
En 2022, I-MAK señalaba que los 10 medicamentos más vendidos en EEUU habían obtenido una media de 74 patentes. Por otro lado, la compañía AbbVie, comercializadora de Humira, el medicamento más caro en 2022, de las 312 solicitudes de patentes sobre el medicamento que se presentaron en la FDA, obtuvo 166.
Además, un estudio sobre los medicamentos que fueron introducidos en el mercado francés entre los años 2006 y 2011, llegó a la conclusión de que el número de moléculas que aportaron un progreso terapéutico importante, disminuyó drásticamente de 22, en el año 2006 a 15, 10, 7, 4 en los años siguientes.
El futuro farmacéutico es ya el presente y no parece alentador. Se resiste una I+D pública.
Detrás de todo este entramado de negocio bursátil se da una problemática que atenta contra el derecho a la salud al impedir el acceso a los medicamentos. Y lo que se consideraba un problema de los países en desarrollo, que ya se opusieron a los Acuerdos de los ADIPC de 1995 por las consecuencias en los precios que acarrearía y que ya Correa anunciaba entonces, ha llegado a ser efecto común a todo el sistema, afectando también a los países desarrollados; sólo era cuestión de tiempo ante un sector que maximiza ganancias como única estrategia.
En 2014, el mundo desarrollado europeo toma conciencia real de la problemática sobre el acceso a los medicamentos al no poder garantizar el acceso al sofosbuvir, medicamento que permitía erradicar la hepatitis C, debido a su alto precio. Ello determina un punto de inflexión que abre definitivamente el, hasta entonces soterrado, debate sobre la necesaria revisión del funcionamiento del sistema farmacéutico europeo.
El Parlamento Europeo toma la iniciativa mediante la elaboración del primer informe que revisa a nivel europeo la problemática del AM en la UE, señala sus disfunciones y eleva propuestas de mejora.
Al mismo tiempo, el Consejo de Ministros de Salud de la UE celebrado en Ámsterdam en julio de 2016, solicita por primera vez un requilibrio del sistema farmacéutico de la UE, haciendo especial hincapié en la necesidad de revisar el impacto de la propiedad intelectual y en la implementación e impacto de los incentivos a la I+D.
Hasta ahora, en la UE, el mercado farmacéutico se ha caracterizado por un alto nivel de regulación para garantizar su libre circulación como mercancía, pero desde el ámbito de la salud, más allá de la seguridad, no ha recibido la misma atención la calidad de la innovación y, menos aún, el aspecto social de garantía de acceso, en términos de disponibilidad, asequibilidad y accesibilidad, dejando al sector empresarial la capacidad de decidir sobre la priorización de las líneas de investigación en base al mercado y estableciendo su precio.
La amenaza de la pandemia contra el Covid19 supuso un impulso en el objetivo de la UE en salvaguardar la salud pública y vino a definir la Unión Europea de la Salud, que englobó el Pilar Social Europeo. El acceso a los medicamentos fue recogido entonces, por primera vez por parte de las instituciones europeas, como uno de los retos en salud y al que se debe dar respuesta, lo que pretende llevar a cabo dentro de la Estrategia Farmacéutica Europea publicada en 2022 por la CE.
Esta Estrategia Farmacéutica Europea vendrá a determinar el funcionamiento del sistema farmacéutico en las próximas décadas; su acierto, o desacierto pueden tener consecuencias económicas, pero también y, especialmente, en términos de salud y bienestar para la ciudadanía europea.
Por otro lado, el siglo XXI es el siglo de la «revolución tecnológica y digital». La salud tampoco puede ni quedará al margen de los desafíos y oportunidades de esta nueva era del desarrollo científico y tecnológico. Sin embargo, este paradigma puede ser integrado como parte de la respuesta a los retos a los que se enfrenta la sanidad o ahondar en un modelo de negocio que genere nuevos obstáculos y dé origen a nuevas desigualdades.
El desarrollo de terapias avanzadas desde el sector público, como se reconoce hoy aquí en la figura de la Dra. Zurita, representa un ejemplo para que los EEMM, las instituciones europeas, los gobiernos nacionales y la sociedad civil se conciencien del papel y oportunidad que, en este contexto de revisión del modelo farmacéutico, brinda una farmacia pública en una nueva era de la medicina, denominada de precisión, para garantizar la sostenibilidad de los SNS, la equidad y cohesión social.
Esta revisión integral de la legislación farmacéutica europea abre una oportunidad única para la reflexión sobre el funcionamiento del modelo de I+D farmacéutico y su reformulación, en su caso. Sin embargo, hasta ahora, las propuestas presentadas por la CE han ido encaminadas hacia una perpetuación e incluso fortalecimiento del sistema vigente, en lo que el Parlamento Europeo ha ahonda aún más.
Especial mención quiero hacer sobre el Espacio Europeo de Datos Sanitarios, incluido dentro del paquete legislativo de la Estrategia Farmacéutica Europea, cuya formulación supone una descapitalización de los sistemas sanitarios públicos con la transferencia altruista al sector privado de la información recopilada a lo largo de décadas y que aumentará de forma exponencial con el desarrollo tecnológico, cuyos resultados derivados la I+D realizada con los mismos los volverá a pagar el propio sistema sanitario público mediante la adquisición de las terapias.
Por qué no desarrollar decididamente un sistema público de investigación que normalice lo llevado a cabo por la Dra. Zurita en el Hospital Puerta de Hierro o Joan Manel en el Hospital Clinic de Barcelona con las CAR-T. De momento, en España, no hemos más que asistido a la puesta en marcha de una iniciativa de empresa público-privada dentro del PERTE de Salud, cuyas condiciones de explotación y beneficios no quedan claras en lo que respecta al sector público.
Desde la AAJM abogamos por una investigación farmacéutica pública y para ellos nos basamos en datos como los publicados por la OCDE sobre 2014 y 2018 que señalan que la financiación pública de la I+D farmacéutica es superior al 40% en la UE; lo que representó en 2014 un 43% de financiación pública respecto de la privada, un 0,14% y un 0,06% del PIB respectivamente, y del 67,5% en 2019, lo que supuso un 0,1% y un 0,07% del PIB respectivamente.
Sin embargo, todo apunta a que no sólo se incentivará una I+D pública para dar respuestas a las necesidades médicas no cubiertas al rechazarse de forma explítica la iniciativa en forma de enmienda firmada por 50 eurodiputados en el Parlamento Europeo, sino que el objetivo principal no será mejorar el acceso a los medicamentos, sino dar respuesta a una supuesta pérdida de competitividad del sector industrial farmacéutico ante la emergencia de países competidores de la región asiática.
Es cierto que hace 25 años 1 de cada dos medicamentos nuevos se originaba en Europa, mientras que ahora es 1 de cada 4 o 5 y que en 2022, en China y en la EU se originaron 18 y 19 nuevos medicamentos, respectivamente. Sin embargo, nada se dice que China, uno de los competidores principales que se asoman al mercado internacional de medicamento, invierte en I+D farmacéutica una décima parte de lo que declara EEUU y una quinta parte que la UE, lo que apuntarían a un sistema más eficiente, a falta de conocer lo que se imputa en ID otras actividades en EEUU y la UE, al mismo tiempo que pareciese que lo que realmente se pretende es mantener un alto nivel de retorno de la inversión, un sector altamente lucrativo.
España
No quiero dejar de referirme a España, pues en nuestro país, el Gobierno, ha abierto revisiones o impulsado normativas respecto de la política farmacéutica que pueden suponer una oportunidad para mejorar el AM y la sostenibilidad de los SNS.
La revisión de la Ley de Garantías y de Uso Racional del Medicamento puede ser una oportunidad para introducir una experiencia piloto de I+D pública, así como para mejorar el uso de medicamentos genéricos y ahorro del gasto farmacéutico eliminando el margen de beneficio destinado a la farmacia, por ejemplo.
La Ley de Equidad para eliminar los copagos y derogar definitivamente los coletazos pendientes del “RD 2012”.
El Reglamento Sobre Terapias Avanzadas (SOHO), recientemente revisado en la UE bajo la presidencia española del Consejo, para la introducción en él de las CAR-T y protegerlas del riesgo de limitar la exención hospitalaria con la que amenaza la Estrategia Farmacéutica Europea Europea y que, además, limitaría el desarrollo público de la ID.
El Real Decreto Sobre Evaluación de Tecnología Sanitaria que propone la transparencia en los costes de los costes de I+D y desarrollo global de los medicamentos pero que debería concretarse hacia su uso en el establecimiento de precios.

Foto de familia de los participantes en el acto de los premios AAJM 2024.
A modo de conclusión
Juan José Rodríguez Sendín, médico de Noblejas durante 36 años, ex presidente de la OMC y uno de los impulsores de la AAJM, puso en valor estos premios con los “queremos impulsar -dijo- la sensibilidad ciudadana en la defensa del medicamento como bien social, un derecho humano preferente al que deben tener acceso todas las personas en condiciones justas” y agradeció al alcalde de Noblejas su implicación en esta causa y en la defensa de la atención sanitaria.
Rodríguez Sendín habló del “incremento de la inequidad social que ocurre -dijo- al limitar la capacidad del sistema sanitario para mejorar la salud”. Y, tras cuestionar los “tratamientos inútiles y tecnologías en casos y en pacientes en los que no están indicadas” que está soportando el Sistema Nacional de Salud por “comportamientos de los Gobiernos sanitarios e intereses contrarios al interés del paciente”, abogó por el deber del médico para “ajustar sus actos y decisiones exclusivamente a la necesidad sanitaria del paciente”.